A number of psychedelic tryptamines, notably N,N-Dimethyltryptamine (DMT), are not orally active because they are rapidly metabolized by the monoamine oxidase (MAO) enzymes in the gut and liver. DMT becomes active only when administered through inhalation, intravenous, intramuscular, or rectal routes, which largely bypass MAO. These methods of administration lead to varied experiences. For example, when inhaled, DMT induces a rapid, intense trip that is markedly different from the introspective ayahuasca journey.
However, these alternative routes are less convenient and consistent compared to taking a pill or tincture. Moreover, the associations of smoke and needles with addictive drugs add a stigma that can create additional barriers to widespread adoption of psychedelics.
DMT can be made orally active when combined with monoamine oxidase inhibitors (MAOIs), such as the harmala alkaloids. These inhibitors work by binding to the active sites of MAO, preventing it from metabolizing DMT. There are two types of MAOIs: reversible MAOIs, which eventually detach from the MAO enzymes, and irreversible MAOIs, which remain bound longer.
Unfortunately, harmala alkaloids can induce nausea. Harmine and harmaline are challenging to synthesize, so they are typically extracted from plant sources. This process requires significant amounts of plant material, and the resulting purity varies.
There is a safe, legal, reversible synthetic MAOI – moclobemide (4-chloro-N-(2-morpholinoethyl)benzamide). It has been around since the 1970s. It is sometimes prescribed as a mild, well-tolerated antidepressant. Below is a simple way to make it, assuming you have a decent vacuum pump (<10 mbar), an argon cylinder, and a short-path distillation apparatus.

Methyl 4-chlorobenzoate (MCB). 30 g 4-chlorobenzoic acid was refluxed in 100 mL methanol with 5 mL 98% sulfuric acid for 6 hours and kept at -30°C overnight. The precipitate was filtered, washed thoroughly with 4°C 100 mL sodium bicarbonate and 2x100 mL water, dried with a desiccant, and distilled under vacuum, yielding 28.12 g (86%) MCB as a white solid with a sweet pine scent, MP 42°C.
4-(2-aminoethyl)morpholine (AEM). 20 g dry 2-chloroethylamine hydrochloride was dissolved in 10 mL water, stirred in 100 mL morpholine for 12 hours, slowly basified with 40 mL 50% sodium hydroxide in an ice bath, stirred vigorously for an hour, and allowed to settle. The organic layer was separated, distilled and dried under vacuum, yielding 17.18 g (77%) AEM as a clear liquid with a faint amine-like smell, MP 24°C.
Moclobemide. 20 g MCB and 15 g AEM were stirred under argon flow at 140°C for 5 hours in an oil bath, letting methanol escape. The mixture was slowly crystallized from 100 mL amyl acetate between 85°C and 4°C. The precipitate was filtered, thoroughly washed with 4°C 80 mL amyl acetate, 2x80 mL heptane, and 3x80 mL water, recrystallized (optionally twice) from 15% ethanol with 4°C 2x80 mL water washes, and dried under vacuum, yielding 12.95 g (42%) moclobemide as a white, odorless solid, MP 138°C.
A second recrystallization from 15% ethanol raises the melting point to 138.5°C. A similar level of purity can also be achieved by drying moclobemide for two days under deep vacuum, at a temperature above the boiling point of AEM, over phosphorus pentoxide, which reacts with any AEM vapors.
Applying several drops of a 0.2% ethanolic ninhydrin solution to a small sample of moclobemide did not result in a purple spot.
For higher sensitivity, more concentrated (e.g. 0.5%) ninhydrin solutions can be used. After a single recrystallization from 15% ethanol the 0.5% ninhydrin test produced a faint purple spot. The 0.5% ninhydrin test was negative after a second recrystallization from 15% ethanol, or after drying moclobemide under deep vacuum at 80°C over phosphorus pentoxide for two days.
The retention factors were measured as follows:

Moclobemide has the following solubility profile:
Unlike other MAOIs, moclobemide is less likely to cause hypertensive crises in the presence of dietary tyramine, but it is still recommended to minimize high-tyramine food and beverages.
As an antidepressant, moclobemide is taken two (150 mg x 2) or three (100 mg x 3) times a day after food. The initial 300 mg daily dose may be increased by 150 mg per week to up to 600 mg daily. Taper over the course of at least several weeks. When switching to a new antidepressant, wait at least 24 hours for moclobemide to wash out.
As an MAOI, 300 mg moclobemide is taken after fasting, 30–60 minutes before DMT. The threshold dose is about 35 mg freebase DMT, 55 mg freebase DMT is the intermediate dose, 75 mg freebase DMT results in a strong trip.
Attach a condenser and heat the mixture until it begins to boil. Run a hose from the top of the condenser outside.
The mixture will become transparent an hour into the reaction, once enough 4-chlorobenzoic acid is consumed. You can monitor the progress by FTIR by watching the characteristic ester C=O peak at ~1723 cm-1.
After 6 hours of refluxing, let the mixture cool down to room temperature, decant it into a beaker, cover it airtight, and place it in the freezer (ideally a deep/medical freezer) overnight.
Quickly filter the methanol off under vacuum. Use ~100 mL ice-cold 5% aqueous sodium bicarbonate solution to wash the crystals in the filter funnel. Wash thoroughly: turn the vacuum off, stir the mass until the slurry is homogeneous, then pull the vacuum again. Tip: use a pipette to wash down the walls of the filter funnel. Use a pH paper to ensure that the filtrate is alkaline. Using the same technique, wash the crystals with ~100 mL cold water, twice. The filtrate should be close to pH-neutral after the final wash.
Dry the crystals over molecular sieves or phosphorus pentoxide at atmospheric pressure over a few days. Drying is recommended as water tends to co-distill with MCB. Drying under vacuum is inconvenient as MCB tends to sublimate.
Purify MCB by short-path vacuum distillation (use a vacuum pump that can produce 10 mbar or deeper vacuum) in an oil bath. Use warm (over 40°C) water to prevent MCB from solidifying inside the condenser. If MCB is overheated and becomes yellow, redistill it more carefully.
Pure MCB is a white solid with a pleasant, sweet scent that resembles pine tar.
To a 250 mL flask add 20 g 2-chloroethylamine hydrochloride and 10 mL distilled water. Stir the mixture until complete dissolution, which is endothermic and takes several minutes. Add 100 mL ice-cold morpholine to the flask. Stopper the flask and gently stir the mixture for 12 hours overnight.
You can monitor the progress by FTIR by watching the characteristic AEM peak at ~1010 cm-1.
Place the flask in an ice bath and let it cool down. Add 40 mL 50% aqueous sodium hydroxide (a saturated NaOH solution will also work) in small portions while stirring vigorously. NaCl will be precipitating out. Continue stirring for 30–60 minutes at room temperature, then let the mixture settle at room temperature.
Carefully separate the top organic layer: decant it through a fast (coffee) filter into a separatory funnel, wash the remaining slurry with a bit of fresh morpholine and decant it through the same filter. Rinse the filter with a bit of fresh morpholine. Discard the bottom aqueous layer. Transfer the organic layer into a 250 mL boiling flask, rinsing the funnels with a bit of fresh morpholine.
Set up for short-path vacuum distillation, preferably in an oil bath. Set the vacuum to the deepest level compatible with your cooling system and distill out the unreacted morpholine first. Set the heating level so that morpholine distills slowly, which ensures that not much AEM will co-distill with it. The vapor temperature will remain steady at first, then rise and drop.
Increase the coolant temperature above the boiling point of morpholine to force it out of the condenser. Gradually increase heating until AEM begins to distill. Use a heat gun to force any morpholine condensed on the walls of the distillation apparatus out. Allow the AEM forerun (a few mL) to distill over to clean the condensation path. Change the receiving flask and collect the remaining AEM; use a heat gun to force the last drops out.
You can use FTIR to detect if distilled AEM is contaminated with water (by a broad elevation around ~3300 cm-1) or morpholine (by elevated IR absorbance at ~3334, ~1093, ~786, and ~593 cm-1). If water contamination is detected (or cannot be ruled out), dry the liquid by keeping it under deep vacuum for several days, at room temperature, in an open flask, over molecular sieves or phosphorus pentoxide.
AEM looks like a clear, colorless liquid that quickly solidifies in the refrigerator, if pure. It has a faint amine-like smell. As a primary amine, AEM reacts with ninhydrin, forming a deep purple complex.
AEM is sensitive to oxidation. It is best to store it in the freezer, melting it right before handling.
Insulate the flask with aluminum foil and heat the mixture as gently as possible, in an oil bath stirred with a secondary small stir bar. Tip: you can use a 1000 mL aluminum heating block as an oil bath container. Once the temperature of the mixture reaches 140°C, maintain it for 5 more hours. The reaction is mildly exothermic with its rate decreasing over time, so continuous temperature monitoring and heating adjustments are necessary.
Traces of oxygen in the argon, high temperatures, and prolonged reaction times all cause moclobemide to degrade. The color of the reaction mixture should remain as clear as possible – yellow, orange, amber, dark red colors indicate progressive stages of degradation.
You can monitor the reaction by observing drops of methanol produced in the reaction. You could also use FTIR by taking samples through a rubber septum and watching the intensity of the characteristic amide N–H peak at ~3276 cm-1.
Under positive argon flow, lift the flask from the oil bath and use a fan to cool the mixture below ~85°C. To remove the unreacted MCB, add 100 mL amyl acetate to the mixture with stirring, and lower the flask into the oil bath to reheat it. All solids should fully dissolve at ~85°C. Transfer the hot solution into a crystallization container, cover it, insulate it well, let it slowly cool to room temperature, and place it in the refrigerator overnight.
Filter the precipitate under vacuum. Add ~80 mL ice-cold amyl acetate to the funnel, quickly stir the residue into a homogeneous slurry, and pull the vacuum to remove the solvent while it is still cold. Repeat the ice-cold wash 2 times with ~80 mL heptane and 3 times ~80 mL water.
To remove more unreacted AEM, dissolve the residue in 15% ethanol (start with ~200 mL and add as necessary) preheated to ~85°C in an oil bath. Once all solids dissolve, slowly cool the mixture to room temperature and place it in the refrigerator overnight. Filter the crystals under vacuum, washing twice with ~80 mL ice-cold water. Tip: when pulling the vacuum, keep the filter funnel covered with a watch glass or petri dish. Collect and dry moclobemide under vacuum.
If extra high purity is required, either perform another recrystallization from 15% ethanol, or dry moclobemide under deep vacuum above the boiling point of AEM, over phosphorus pentoxide, for several days. Phosphorus pentoxide will react with any AEM vapors, turning yellow.
Store moclobemide at room temperature (below 40°C) in a dry, dark place. For greater stability, it can be converted to its hydrochloride salt.
However, these alternative routes are less convenient and consistent compared to taking a pill or tincture. Moreover, the associations of smoke and needles with addictive drugs add a stigma that can create additional barriers to widespread adoption of psychedelics.
DMT can be made orally active when combined with monoamine oxidase inhibitors (MAOIs), such as the harmala alkaloids. These inhibitors work by binding to the active sites of MAO, preventing it from metabolizing DMT. There are two types of MAOIs: reversible MAOIs, which eventually detach from the MAO enzymes, and irreversible MAOIs, which remain bound longer.
Unfortunately, harmala alkaloids can induce nausea. Harmine and harmaline are challenging to synthesize, so they are typically extracted from plant sources. This process requires significant amounts of plant material, and the resulting purity varies.
There is a safe, legal, reversible synthetic MAOI – moclobemide (4-chloro-N-(2-morpholinoethyl)benzamide). It has been around since the 1970s. It is sometimes prescribed as a mild, well-tolerated antidepressant. Below is a simple way to make it, assuming you have a decent vacuum pump (<10 mbar), an argon cylinder, and a short-path distillation apparatus.
Synthesis
Methyl 4-chlorobenzoate (MCB). 30 g 4-chlorobenzoic acid was refluxed in 100 mL methanol with 5 mL 98% sulfuric acid for 6 hours and kept at -30°C overnight. The precipitate was filtered, washed thoroughly with 4°C 100 mL sodium bicarbonate and 2x100 mL water, dried with a desiccant, and distilled under vacuum, yielding 28.12 g (86%) MCB as a white solid with a sweet pine scent, MP 42°C.
4-(2-aminoethyl)morpholine (AEM). 20 g dry 2-chloroethylamine hydrochloride was dissolved in 10 mL water, stirred in 100 mL morpholine for 12 hours, slowly basified with 40 mL 50% sodium hydroxide in an ice bath, stirred vigorously for an hour, and allowed to settle. The organic layer was separated, distilled and dried under vacuum, yielding 17.18 g (77%) AEM as a clear liquid with a faint amine-like smell, MP 24°C.
Moclobemide. 20 g MCB and 15 g AEM were stirred under argon flow at 140°C for 5 hours in an oil bath, letting methanol escape. The mixture was slowly crystallized from 100 mL amyl acetate between 85°C and 4°C. The precipitate was filtered, thoroughly washed with 4°C 80 mL amyl acetate, 2x80 mL heptane, and 3x80 mL water, recrystallized (optionally twice) from 15% ethanol with 4°C 2x80 mL water washes, and dried under vacuum, yielding 12.95 g (42%) moclobemide as a white, odorless solid, MP 138°C.
Characterization
Melting point
Moclobemide, after a single recrystallization from 15% ethanol and drying, melts around 138.3°C, which matches the literature value of 135-138°C.A second recrystallization from 15% ethanol raises the melting point to 138.5°C. A similar level of purity can also be achieved by drying moclobemide for two days under deep vacuum, at a temperature above the boiling point of AEM, over phosphorus pentoxide, which reacts with any AEM vapors.
Ninhydrin test
Ninhydrin reacts with primary amines, producing intense purple color. The test is sensitive enough to detect amine impurities in microgram quantities.Applying several drops of a 0.2% ethanolic ninhydrin solution to a small sample of moclobemide did not result in a purple spot.
For higher sensitivity, more concentrated (e.g. 0.5%) ninhydrin solutions can be used. After a single recrystallization from 15% ethanol the 0.5% ninhydrin test produced a faint purple spot. The 0.5% ninhydrin test was negative after a second recrystallization from 15% ethanol, or after drying moclobemide under deep vacuum at 80°C over phosphorus pentoxide for two days.
TLC
75 mg MCB, AEM and moclobemide were dissolved in 25 mL MeOH each and developed on a silica gel 60 Å F254 TLC plate using the DCM:EtOH 10:1 (v/v) eluent system. The moclobemide and MCB spots were visible under 254 nm UV light, while a drop of 0.2% ethanolic ninhydrin was used to visualize AEM.The retention factors were measured as follows:
- MCB Rf ≈ 0.83
- AEM Rf ≈ 0.00 (does not move)
- Moclobemide Rf ≈ 0.25
IR
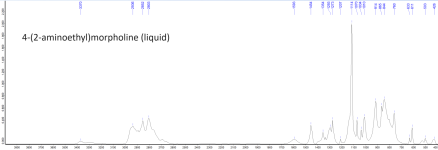
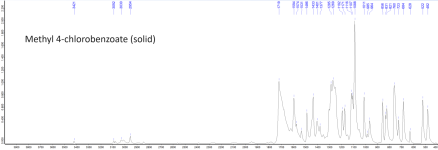
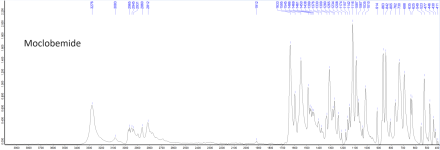
Attached below are the ATR FTIR absorbance spectra of MCB (solid), AEM (molten), and moclobemide with baseline correction, min-max normalization, and peak peaking applied. The raw uncorrected spectra are also attached as data tables.Solubility
Moclobemide, a tertiary amine, forms water-soluble salts with acids. Moclobemide acetate is more soluble in water than moclobemide hydrochloride. The hydrochloride salt can be obtained by dissolving moclobemide in dilute acetic acid, saturating the solution with NaCl, filtering the precipitate and recrystallizing it from MeOH. The salt crystallizes in the freezer very slowly.Moclobemide has the following solubility profile:
- highly soluble in chloroform (33.6g/100mL), THF, MeOH (11.8g / 100 mL)
- soluble in DMSO (5.4 g / 100mL), EtOH (2.69 g / 100 mL), IPA, DCM, EtOAc (1 g / 45 mL), AmOAc (1 g / 100 mL)
- poorly soluble in water (0.4 g / 100 mL), heptane, diethyl ether
Contraindications and Dosage
Avoid medications like slegiline, meperidine (pethidine), thioridazine, dextromethorphan (in many cough medicines), bupropion, triptans, and tramadol.Unlike other MAOIs, moclobemide is less likely to cause hypertensive crises in the presence of dietary tyramine, but it is still recommended to minimize high-tyramine food and beverages.
As an antidepressant, moclobemide is taken two (150 mg x 2) or three (100 mg x 3) times a day after food. The initial 300 mg daily dose may be increased by 150 mg per week to up to 600 mg daily. Taper over the course of at least several weeks. When switching to a new antidepressant, wait at least 24 hours for moclobemide to wash out.
As an MAOI, 300 mg moclobemide is taken after fasting, 30–60 minutes before DMT. The threshold dose is about 35 mg freebase DMT, 55 mg freebase DMT is the intermediate dose, 75 mg freebase DMT results in a strong trip.
Bioassay
A trial involving 300 mg of moclobemide in a dilute citric acid solution showed no significant initial effects and no nausea. Thirty minutes later, 80 mg of DMT benzoate (equivalent to 48.5 mg freebase DMT) was administered, leading to a noticeable psychedelic experience starting 30 minutes after ingestion. The experience was marked by mild nausea, vasoconstriction, yawning, and significant closed-eye visuals, but no open-eye visuals. The peak was followed by a calm, euphoric plateau lasting 2 hours, with afterglow effects dissipating over the following 2 hours.Detailed Synthesis Instructions
MCB
Add 100 mL ice-cold methanol to a 250 mL boiling flask. With vigorous stirring, add 5 mL concentrated sulfuric acid dropwise, followed by 30 g 4-chlorobenzoic acid.Attach a condenser and heat the mixture until it begins to boil. Run a hose from the top of the condenser outside.
The mixture will become transparent an hour into the reaction, once enough 4-chlorobenzoic acid is consumed. You can monitor the progress by FTIR by watching the characteristic ester C=O peak at ~1723 cm-1.
After 6 hours of refluxing, let the mixture cool down to room temperature, decant it into a beaker, cover it airtight, and place it in the freezer (ideally a deep/medical freezer) overnight.
Quickly filter the methanol off under vacuum. Use ~100 mL ice-cold 5% aqueous sodium bicarbonate solution to wash the crystals in the filter funnel. Wash thoroughly: turn the vacuum off, stir the mass until the slurry is homogeneous, then pull the vacuum again. Tip: use a pipette to wash down the walls of the filter funnel. Use a pH paper to ensure that the filtrate is alkaline. Using the same technique, wash the crystals with ~100 mL cold water, twice. The filtrate should be close to pH-neutral after the final wash.
Dry the crystals over molecular sieves or phosphorus pentoxide at atmospheric pressure over a few days. Drying is recommended as water tends to co-distill with MCB. Drying under vacuum is inconvenient as MCB tends to sublimate.
Purify MCB by short-path vacuum distillation (use a vacuum pump that can produce 10 mbar or deeper vacuum) in an oil bath. Use warm (over 40°C) water to prevent MCB from solidifying inside the condenser. If MCB is overheated and becomes yellow, redistill it more carefully.
Pure MCB is a white solid with a pleasant, sweet scent that resembles pine tar.
AEM
For consistent yields, dry 2-chloroethylamine hydrochloride in a vacuum oven at ~50°C over molecular sieves or phosphorus pentoxide immediately before the reaction, as it is quite hygroscopic.To a 250 mL flask add 20 g 2-chloroethylamine hydrochloride and 10 mL distilled water. Stir the mixture until complete dissolution, which is endothermic and takes several minutes. Add 100 mL ice-cold morpholine to the flask. Stopper the flask and gently stir the mixture for 12 hours overnight.
You can monitor the progress by FTIR by watching the characteristic AEM peak at ~1010 cm-1.
Place the flask in an ice bath and let it cool down. Add 40 mL 50% aqueous sodium hydroxide (a saturated NaOH solution will also work) in small portions while stirring vigorously. NaCl will be precipitating out. Continue stirring for 30–60 minutes at room temperature, then let the mixture settle at room temperature.
Carefully separate the top organic layer: decant it through a fast (coffee) filter into a separatory funnel, wash the remaining slurry with a bit of fresh morpholine and decant it through the same filter. Rinse the filter with a bit of fresh morpholine. Discard the bottom aqueous layer. Transfer the organic layer into a 250 mL boiling flask, rinsing the funnels with a bit of fresh morpholine.
Set up for short-path vacuum distillation, preferably in an oil bath. Set the vacuum to the deepest level compatible with your cooling system and distill out the unreacted morpholine first. Set the heating level so that morpholine distills slowly, which ensures that not much AEM will co-distill with it. The vapor temperature will remain steady at first, then rise and drop.
Increase the coolant temperature above the boiling point of morpholine to force it out of the condenser. Gradually increase heating until AEM begins to distill. Use a heat gun to force any morpholine condensed on the walls of the distillation apparatus out. Allow the AEM forerun (a few mL) to distill over to clean the condensation path. Change the receiving flask and collect the remaining AEM; use a heat gun to force the last drops out.
You can use FTIR to detect if distilled AEM is contaminated with water (by a broad elevation around ~3300 cm-1) or morpholine (by elevated IR absorbance at ~3334, ~1093, ~786, and ~593 cm-1). If water contamination is detected (or cannot be ruled out), dry the liquid by keeping it under deep vacuum for several days, at room temperature, in an open flask, over molecular sieves or phosphorus pentoxide.
AEM looks like a clear, colorless liquid that quickly solidifies in the refrigerator, if pure. It has a faint amine-like smell. As a primary amine, AEM reacts with ninhydrin, forming a deep purple complex.
AEM is sensitive to oxidation. It is best to store it in the freezer, melting it right before handling.
Moclobemide
Equip a 250 mL boiling flask with a thermocouple and a gas inlet-outlet adapter connected to an argon tank and a gas bubbler. Set up for short-path distillation to allow methanol to escape from the reaction mixture; there is no need to cool the condenser. Add 20 g MCB and 15 g AEM to the flask and slowly flush it with argon a few times. Before heating begins, set up constant argon flow and maintain it during the entire reaction and cooldown.Insulate the flask with aluminum foil and heat the mixture as gently as possible, in an oil bath stirred with a secondary small stir bar. Tip: you can use a 1000 mL aluminum heating block as an oil bath container. Once the temperature of the mixture reaches 140°C, maintain it for 5 more hours. The reaction is mildly exothermic with its rate decreasing over time, so continuous temperature monitoring and heating adjustments are necessary.
Traces of oxygen in the argon, high temperatures, and prolonged reaction times all cause moclobemide to degrade. The color of the reaction mixture should remain as clear as possible – yellow, orange, amber, dark red colors indicate progressive stages of degradation.
You can monitor the reaction by observing drops of methanol produced in the reaction. You could also use FTIR by taking samples through a rubber septum and watching the intensity of the characteristic amide N–H peak at ~3276 cm-1.
Under positive argon flow, lift the flask from the oil bath and use a fan to cool the mixture below ~85°C. To remove the unreacted MCB, add 100 mL amyl acetate to the mixture with stirring, and lower the flask into the oil bath to reheat it. All solids should fully dissolve at ~85°C. Transfer the hot solution into a crystallization container, cover it, insulate it well, let it slowly cool to room temperature, and place it in the refrigerator overnight.
Filter the precipitate under vacuum. Add ~80 mL ice-cold amyl acetate to the funnel, quickly stir the residue into a homogeneous slurry, and pull the vacuum to remove the solvent while it is still cold. Repeat the ice-cold wash 2 times with ~80 mL heptane and 3 times ~80 mL water.
To remove more unreacted AEM, dissolve the residue in 15% ethanol (start with ~200 mL and add as necessary) preheated to ~85°C in an oil bath. Once all solids dissolve, slowly cool the mixture to room temperature and place it in the refrigerator overnight. Filter the crystals under vacuum, washing twice with ~80 mL ice-cold water. Tip: when pulling the vacuum, keep the filter funnel covered with a watch glass or petri dish. Collect and dry moclobemide under vacuum.
If extra high purity is required, either perform another recrystallization from 15% ethanol, or dry moclobemide under deep vacuum above the boiling point of AEM, over phosphorus pentoxide, for several days. Phosphorus pentoxide will react with any AEM vapors, turning yellow.
Store moclobemide at room temperature (below 40°C) in a dry, dark place. For greater stability, it can be converted to its hydrochloride salt.
Attachments
Last edited:
 No need for you to try this, of course, since you clearly have the 'real thing'.
No need for you to try this, of course, since you clearly have the 'real thing'.