
SWIM had a dream.
An extract of peganum harmala was made with acetic acid and water. It was basified with sodium carbonate and salt. After precipitation a brown residue was obtained. This was recrystallized once in ethanol. After brown crystals formed they were filtered. The supernatant was dried. It was red. Swim expects this is what people commonly refer to as harmala red?
SWIM analyzed this material by LC-MS using APCI probe operating in positive ion mode. UV measured at 254nm and 380nm. C18 column methanol water gradient...details not necessary at this point.
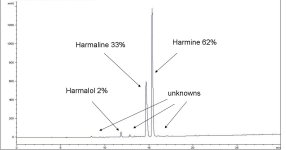
The results were mostly harmine and harmaline. As well as some harmalol and some unknown compounds.
SWIM has not analyzed the initial material. Next time when SWIM has time to dream SWIM will analyze the initial extract, the recrystallized material and the supernatant. To determine a bit more whats going on.
Note SWIM has also analyzed this material by GC and its also mostly harmine and harmaline SWIM can't remember if harmalol was there.
Anyway it seems yet again what we thought was going on: degradation, or some major impurity is unlikely. But more experiments need to be done. I post a UV chromatogram with relative % abundance of major 3 harmaloids.
An extract of peganum harmala was made with acetic acid and water. It was basified with sodium carbonate and salt. After precipitation a brown residue was obtained. This was recrystallized once in ethanol. After brown crystals formed they were filtered. The supernatant was dried. It was red. Swim expects this is what people commonly refer to as harmala red?
SWIM analyzed this material by LC-MS using APCI probe operating in positive ion mode. UV measured at 254nm and 380nm. C18 column methanol water gradient...details not necessary at this point.
The results were mostly harmine and harmaline. As well as some harmalol and some unknown compounds.
SWIM has not analyzed the initial material. Next time when SWIM has time to dream SWIM will analyze the initial extract, the recrystallized material and the supernatant. To determine a bit more whats going on.
Note SWIM has also analyzed this material by GC and its also mostly harmine and harmaline SWIM can't remember if harmalol was there.
Anyway it seems yet again what we thought was going on: degradation, or some major impurity is unlikely. But more experiments need to be done. I post a UV chromatogram with relative % abundance of major 3 harmaloids.
")