
Samvidbuho
Esteemed member
Howdy Friends,
Been a while since I posted. I was running some thermal analysis on a recent batch and noticed some preeetty interesting behavior I thought I'd share with you all.
This last batch (Max Purity: 99.19% ) demonstrated something I thought was strange on the DSC thermograms. I have been producing crystals (heh - with difficulty, out of amorphous extract) with a characteristic melting point [Tm(1) 68.9-69.8] similar to that described by Shulgin and Shulgin [Tm 67-68] (The amorphous starting material melts at ~58C). On the thermograms this is indicated by the large endotherm seen in the first heating cycle
) demonstrated something I thought was strange on the DSC thermograms. I have been producing crystals (heh - with difficulty, out of amorphous extract) with a characteristic melting point [Tm(1) 68.9-69.8] similar to that described by Shulgin and Shulgin [Tm 67-68] (The amorphous starting material melts at ~58C). On the thermograms this is indicated by the large endotherm seen in the first heating cycle
During this first heating cycle, the crystalline material (Samples 1,2,4) exhibits a single melting peak at ~69.65C, and absence of exothermic event on the cooling.
During the 2nd heating cycle things get interesting - Sample 1 demonstrates double melting peaks without an intermediate cold crystallization peak at Tm(2): ~44-46C & Tm(3): ~56-58C, whereas Sample 2 demonstrates a single melting peak at 46C, without any apparent recrystallization peak anywhere.
Sample 1 was a single crystal, 8.4mg, (TZ Alum pan, crimped) which actually demonstrated some amorphous residue on the outside of the pan afte ranalysis
Sample 2 was very fine crystals which were recrystallized again, 5.27mg, no residue after.
Heating 1 was performed at 1K/min, Cooling at 2.5K/min, and 2nd Heating at 1K/min
Temperature cycle was (RT) to 100C, isotherm 2 min, ramp to -15C, hold 2 min, then ramp to 100C.
There is a lot to be said, but it appears to me:
1. 3 Different polymorphic forms of DMT are clearly evident (as described elsewhere)
2. As seen during reheating, there seems to be a polymorphic transition that occurs as the 'Form A' is actually melting. The melting of 'Form B' then kind of balances any exothermal events during this transition. Any other thoughts?
3. An interesting observation is that all of the endothermic peaks seen during the reheating cycles are extremely high purity, >99.95% in most cases. I wonder what is happening after the original crystal melts? Obviously the higher MP form is the more thermodynamically stable, but it looks like the energy barrier to nucleate again is very high?
Thought everyone would find this interesting as well.
Attached are all the DSC thermograms for 4 Samples.
Sample 1: Crystalline, 2 melting peaks on 2nd heating
Sample 2: Crystalline, 99.19% Purity, single melting peak on reheat
Sample 3: Amorphous, exothermal peak on cooling with no subsequent thermal events
Sample 4: Crystalline, single melting peak on reheat but also with preceding exotherm
At the end is a reprint of mine of various literature values for DMT MPs.
Be very well,
Been a while since I posted. I was running some thermal analysis on a recent batch and noticed some preeetty interesting behavior I thought I'd share with you all.
This last batch (Max Purity: 99.19%
) demonstrated something I thought was strange on the DSC thermograms. I have been producing crystals (heh - with difficulty, out of amorphous extract) with a characteristic melting point [Tm(1) 68.9-69.8] similar to that described by Shulgin and Shulgin [Tm 67-68] (The amorphous starting material melts at ~58C). On the thermograms this is indicated by the large endotherm seen in the first heating cycleDuring this first heating cycle, the crystalline material (Samples 1,2,4) exhibits a single melting peak at ~69.65C, and absence of exothermic event on the cooling.
During the 2nd heating cycle things get interesting - Sample 1 demonstrates double melting peaks without an intermediate cold crystallization peak at Tm(2): ~44-46C & Tm(3): ~56-58C, whereas Sample 2 demonstrates a single melting peak at 46C, without any apparent recrystallization peak anywhere.
Sample 1 was a single crystal, 8.4mg, (TZ Alum pan, crimped) which actually demonstrated some amorphous residue on the outside of the pan afte ranalysis
Sample 2 was very fine crystals which were recrystallized again, 5.27mg, no residue after.
Heating 1 was performed at 1K/min, Cooling at 2.5K/min, and 2nd Heating at 1K/min
Temperature cycle was (RT) to 100C, isotherm 2 min, ramp to -15C, hold 2 min, then ramp to 100C.
There is a lot to be said, but it appears to me:
1. 3 Different polymorphic forms of DMT are clearly evident (as described elsewhere)
2. As seen during reheating, there seems to be a polymorphic transition that occurs as the 'Form A' is actually melting. The melting of 'Form B' then kind of balances any exothermal events during this transition. Any other thoughts?
3. An interesting observation is that all of the endothermic peaks seen during the reheating cycles are extremely high purity, >99.95% in most cases. I wonder what is happening after the original crystal melts? Obviously the higher MP form is the more thermodynamically stable, but it looks like the energy barrier to nucleate again is very high?
Thought everyone would find this interesting as well.
Attached are all the DSC thermograms for 4 Samples.
Sample 1: Crystalline, 2 melting peaks on 2nd heating
Sample 2: Crystalline, 99.19% Purity, single melting peak on reheat
Sample 3: Amorphous, exothermal peak on cooling with no subsequent thermal events
Sample 4: Crystalline, single melting peak on reheat but also with preceding exotherm
At the end is a reprint of mine of various literature values for DMT MPs.
Be very well,
Attachments
-
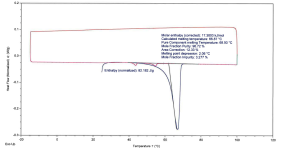 Sample+1%2C+DSC+Thermogram+(with+Purity+Analysis).png186.8 KB · Views: 2
Sample+1%2C+DSC+Thermogram+(with+Purity+Analysis).png186.8 KB · Views: 2 -
Thermal+Analysis+of+Polymorphic+Phase+Transitions+in+DMT.pdf726 KB · Views: 1
-
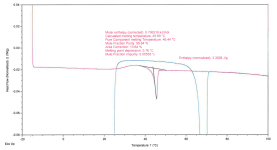 Sample+2%2C+Close+Up+of+Single+Exo-Endothermal+Event+During+Reheating+(Focus+Endotherm).png156.4 KB · Views: 3
Sample+2%2C+Close+Up+of+Single+Exo-Endothermal+Event+During+Reheating+(Focus+Endotherm).png156.4 KB · Views: 3 -
 Sample+1%2C+Close+Up+of+Double+Melting+Peaks+During+2nd+Heating+Cycle.png248.3 KB · Views: 3
Sample+1%2C+Close+Up+of+Double+Melting+Peaks+During+2nd+Heating+Cycle.png248.3 KB · Views: 3 -
 DSC+Thermogram+Comparison+to+Previous+Lot%2C+Showing+Single+Melting+Peak+Preceded+by+Exotherma...png210.5 KB · Views: 3
DSC+Thermogram+Comparison+to+Previous+Lot%2C+Showing+Single+Melting+Peak+Preceded+by+Exotherma...png210.5 KB · Views: 3 -
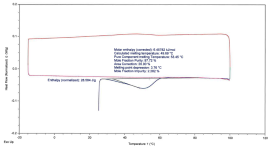 Sample+3+(Amorphous)%2C+DSC+Thermogram+of+HCH+Cycle+Showing+Melting+Peak+on+First+Heating.png111.9 KB · Views: 2
Sample+3+(Amorphous)%2C+DSC+Thermogram+of+HCH+Cycle+Showing+Melting+Peak+on+First+Heating.png111.9 KB · Views: 2 -
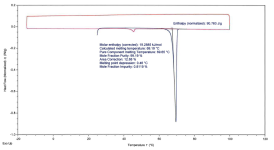 Sample+2%2C+DSC+Thermogram+(with+Purity+Analysis).png103.9 KB · Views: 2
Sample+2%2C+DSC+Thermogram+(with+Purity+Analysis).png103.9 KB · Views: 2
")




