So I was following a minor modification of the VDS harmaline reduction tek.
All I did differently was to use a magnetic stir plate and keep the reaction warm so it went faster, and my vinegar is 5%. I opted for the harmine/harmaline combo reduction with subsequent separation by means of bicarbonate/carbonate.
All went well. I got a big light tan ball of harmine and zinc carbonate, and a somewhat darker tan ball of THH freebase. I dissolved each in its own moderate excess of vinegar and proceeded to manske them by heating them, dissolving in salt, cooling a bit, and refrigerating.
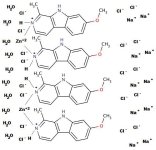
When the salt dissolved in the harmine solution it formed that red oil that was observed in the THH project thread when someone tried to directly manske the filtered reduction solution.
I filtered it and more formed. All beakers are in the fridge for the night now.
Has any more been discovered about this oil? Particularly what to do next?...
My current plan is to wait for tomorrow and if I can get crystals out I'll do another manske on them. I'm toying with the idea of including a whopping dose of ammonium sulfate to distract any zinc that may be grabbing on to my alkaloids.
Unvarnished lab notes:
Thanks")
All I did differently was to use a magnetic stir plate and keep the reaction warm so it went faster, and my vinegar is 5%. I opted for the harmine/harmaline combo reduction with subsequent separation by means of bicarbonate/carbonate.
All went well. I got a big light tan ball of harmine and zinc carbonate, and a somewhat darker tan ball of THH freebase. I dissolved each in its own moderate excess of vinegar and proceeded to manske them by heating them, dissolving in salt, cooling a bit, and refrigerating.
When the salt dissolved in the harmine solution it formed that red oil that was observed in the THH project thread when someone tried to directly manske the filtered reduction solution.
I filtered it and more formed. All beakers are in the fridge for the night now.
Has any more been discovered about this oil? Particularly what to do next?...
My current plan is to wait for tomorrow and if I can get crystals out I'll do another manske on them. I'm toying with the idea of including a whopping dose of ammonium sulfate to distract any zinc that may be grabbing on to my alkaloids.
Unvarnished lab notes:
500 grams of P. harmala extracted and 5 successive salt crystallizations performed
0.12 grams of harm(al)ine HCl set aside as reference
400 ml distilled vinegar [5% acidity] heated to scalding, 25.5 grams of harm(al)ine HCl dissolved into it in a flat bottom boiling flask
Solution placed on unheated magnetic stir plate and 12 grams of ultra-pure zinc powder added
Mixture turned milky greenish grey and maintained a gentle vortex
T+2 hrs much of the zinc has dissolved, mixture has now cooled to 37°C, an additional 3 grams of zinc added
T+3.5 hrs mixture has cooled to room temp and reaction has slowed, reaction flask re-heated to 75°C in a hot water bath and returned to the stir plate, fizzing has resumed
T+6 hrs much of the zinc has dissolved, mixture has cooled to 35°C, mixture was repeatedly decanted and filtered to remove the remaining zinc powder. Solution was placed in a 1 L beaker on the stir plate and 35.45 grams of NaHCO3 was slowly stirred in. After 20 grams the
mixture became too thick to stir smoothly, 100 ml distilled water added. pH measured at ~8. Left to stir for 60 minutes.
Mixture filtered to yield the crude harmine freebase fraction. With stirring the filtered liquid was basified with 10 grams Na2CO3 (anh.)
and filtered to yield the crude THH freebase fraction.
The two fractions were separately dissolved in vinegar and subject to a standard salt crystallization procedure.
The harmine fraction, upon addition of salt, promptly became cloudy. 50 ml of vinegar were added. A red oil formed which adhered to the
beakers. It was filtered and new red oil was formed in the new beakers. The mixture was cooled and moved to the fridge.
The THH fraction behaved normally at first. Upon cooling a yellow precipitate formed and traces of oil were seen. It was moved to the
fridge.
0.12 grams of harm(al)ine HCl set aside as reference
400 ml distilled vinegar [5% acidity] heated to scalding, 25.5 grams of harm(al)ine HCl dissolved into it in a flat bottom boiling flask
Solution placed on unheated magnetic stir plate and 12 grams of ultra-pure zinc powder added
Mixture turned milky greenish grey and maintained a gentle vortex
T+2 hrs much of the zinc has dissolved, mixture has now cooled to 37°C, an additional 3 grams of zinc added
T+3.5 hrs mixture has cooled to room temp and reaction has slowed, reaction flask re-heated to 75°C in a hot water bath and returned to the stir plate, fizzing has resumed
T+6 hrs much of the zinc has dissolved, mixture has cooled to 35°C, mixture was repeatedly decanted and filtered to remove the remaining zinc powder. Solution was placed in a 1 L beaker on the stir plate and 35.45 grams of NaHCO3 was slowly stirred in. After 20 grams the
mixture became too thick to stir smoothly, 100 ml distilled water added. pH measured at ~8. Left to stir for 60 minutes.
Mixture filtered to yield the crude harmine freebase fraction. With stirring the filtered liquid was basified with 10 grams Na2CO3 (anh.)
and filtered to yield the crude THH freebase fraction.
The two fractions were separately dissolved in vinegar and subject to a standard salt crystallization procedure.
The harmine fraction, upon addition of salt, promptly became cloudy. 50 ml of vinegar were added. A red oil formed which adhered to the
beakers. It was filtered and new red oil was formed in the new beakers. The mixture was cooled and moved to the fridge.
The THH fraction behaved normally at first. Upon cooling a yellow precipitate formed and traces of oil were seen. It was moved to the
fridge.
Thanks