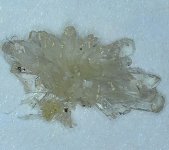
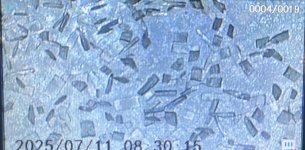
while tidying a spare room i noticed i had left one of my glass containers in there which i use for the freeze process.
it would have certainly appeared empty when i left it there a couple weeks ago i expect.
when i looked in it i found these amazing looking crystals.
im still somewhat new to extracting and have not come across this style of dmt before.
weighs 20mg
it would have certainly appeared empty when i left it there a couple weeks ago i expect.
when i looked in it i found these amazing looking crystals.
im still somewhat new to extracting and have not come across this style of dmt before.
weighs 20mg
Attachments



Last edited: