Aways back I started a post about testing reused solvent. I discovered that the washing method, imo, needed a little more specification, so because it took some time to work out the method i’ll use, for now at least, I moved the post up to be a little more convenient.
Here is how I do the Sodium Carbonate washing:
1. Fill a jar 2/3 - 3/4 full with used solvent. The headspace is needed to accommodate the volume of the wash solution, as well as, for potential expansion of frozen water-precipitate puck.
2. Determine the volume of used solvent. I do this by filling the same type jar that the used solvent is in with tap water, to the same level as the surface of the used solvent; then I pour the water into a graduated cylinder/measuring cup to determine the volume. [I don’t directly measure the volume of the used solvent, because it dissolves the inked markings on my cylinders.]
3. Do the the following calculations, based on the instructions in the wiki:
a. Total volume (mL) needed of sodium carbonate solution = volume of used solvent x 0.20
b. Amounts of sodium carbonate solution ingredients:
i. Grams of sodium carbonate needed = Total volune of sodium carbonate solution needed x 0.35
Ii. Put grams of sodium carbonate needed in mixing jar, add distilled water to equal total volume of sodium carbonate-water solution needed.
4. Mix sodium carbonate and distilled water for 1 minute. Solution will become noticeably warm, sodium carbonate will not fully dissolve.
5. Pour sodium carbonate solution into used solvent, stir well for 1 minute; cover jar but do not screw on lid, as there may be off-gassing of evolving carbon dioxide. Three layers will form: bottom, water layer; middle, sodium carbonate-water layer; top, solvent layer. Initially the solvent will be clear and 2 to 4pH. Over a 20-minute period, mix the solution 3-4 times, 1 min each, to breakup & redistribute the sodium carbonate. The resulting solvent layer should become cloudy and pH will move closer to 5 (pH of fresh EA is solid 5ish). Screw on jar lid, and let solution clear as best possible, for approximately 1 week..
6. After most of the cloudiness has dissipated, place jar in freezer for 24 hours.
7. After freezing solvent for 24 hours, decant solvent through a stainless steel sieve into a coffee filter to remove any floating ice crystals and other residues. As soon as all the floating ice has been collected in the sieve, remove it from above the coffee filter so that you minimize the opportunity for melted ice to reintroduce water back into the solvent. After the solvent is filtered, put lid on collection jar, and store at room temperature until the cloudiness settles and solvent becomes clear. A haze is likely to be present even after the solvent is no longer cloudy. When you shine a flashlight through the solvent, if it looks clear but you can’t make out the details of the flashlight, you still have a haze of particles that will eventually settle out. I want my solvent to be as clear as possible, because I don’t want this crap ending up in my product, so I’m willing to wait however long it takes to fully clear, which could be a week or more. Once at a satisfactory level of clarity, decant the clear solvent off of the settled particles, and store for future use. You may collect the remaining cloudy solvent/sludge in a smaller container, letting it settle until the solvent clears and then decant/syphon that off, to increase the amount of solvent recovery.
For those with Buchner funnels, I have some Whatman qualitative filters ordered, #5 and 602H, to see how well they do at reducing the haze. This doesn’t work, cloudiness/haze particles too small.
Did a new sodium carbonate wash (steps 1- 5 above) on 3 jars of used standard run solvent, and 4 used “yellow” solvents (from boiled water std paste, microwaved paste, chilled paste, microwaved & chilled paste runs). See Yellow Solvent pic below. 2 of the 3 used standard solvent jars were then treated as described here: Ethyl acetate approach [CIELO] - Preparation - Welcome to the DMT-Nexus These treatments will then be used in a second attempt/version of part I below. The other 5 jars will be treated with the “best” washing method from this trial, then used in part II below.
See pics below “2nd Approach 72 Hrs After Washing” and “2nd Approach 72Hrs Bottom”; showing the washed solvent at the 72 hr mark, very difficult to get a good pic, but the separation between the top 2 layers of the solvent is evident (about 5” in total; 40% clear upper, 60% cloudy lower). There are two water layers at the bottom of the jars (about 1” combined); the lowest layer is a heavily saturated with suspended sodium carbonate crystals, the layer above it looks like pure water. The boundary between the top water layer and the bottom solvent layer has a very thin layer of colored particulate.
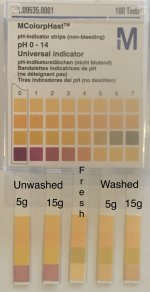
I. Experiment comparing fresh solvent, washed & chilled used solvent (@2 citrate levels from previous use: equivalent of 5 and 15% of material wt), and unwashed used solvent (@2 citrate levels from previous use: 5 and 15% of material wt); using individually prepared 15g runs of standard paste; 6 pulls, 1st one 35mL, 2-6 25mL; 0.75g citric, passive crystallization.
Attached are views of the solvents and their pH strips. All the solvents were crystal clear, the unwashed solvents had visibly darker hue than the washed solvents.
The unwashed pulls are nearly worthless. The 15% citric unwashed pulls were heavily sedimented in each pull, hardly draining through the filter by the end of pulling; the 5% citric unwashed pull was also heavily sedimented, but fully drained through the filter. Additionally, both unwashed solvent pulls became sticky, snot-like, and unstiirable, the 15% citric starting with the 4th pull, the 5% citric with its 6th pull. Both runs. even after multiple filterings and several days of settling, remained so cloudy that light beams from a high intensity flashlight were not visible through the cloudiness. I tried to rehab these with ultrafiltration, with no improvement at 2 mucron pore size. Washes were salted despite cloudiness, 15g equivalent produced a pure white mixture of mescaline citrate needles and what is brlieved to be calcium citrate. Total yield was 2.9%, at least 1/3 if this is suspected to be calcium citrate. The 5g citrate equiv run produced 2.5% yield, about 1/4 of this suspected to be calcium citrate. Use of unwashed solvent is not recommended. Wash & clear your used solvent before running it again.
The fresh solvent and both the washed 5% and 15% solvents ran normally. The final yields were 0.215g (1.4%), 0.296g (2.0%), and 0.379g (2.5%) for fresh, washed 5g, and washed 15g solvent runs, respectively - the exact opposite order of what I expected! The products look the same, 100% needles; and taste the same; and are not mislabeled. Doing jar wash-evap; might be a few more crystals each, but i don’t expect any dramatic changes. Maybe i messed up somehow, but I doubt it. A headscratcher,
Ran another version of this experiment, with the second washing method described in bold above, see Ethyl acetate approach [CIELO] - Preparation - Welcome to the DMT-Nexus
II. Experiment comparing multiple re-uses of solvent from the various treatments that were previously listed as options on the wiki. We know that those treatments reduced yield when using fresh solvent, now I want to see how product quantity & quality, as well as useful solvent life, compare with repeated solvent re-use, using the same treatments: standard paste, boiled water paste, microwaved paste, chilled paste and solvent, microwaved paste with chilled paste and solvent.
The details of this experiment can be found here:
Ethyl acetate approach [CIELO] - Preparation - Welcome to the DMT-Nexus