Adding the the sulfate salt while water is basic (no common ion effect) could precipitate gunk but not mescaline. After that, neutralize (or go to ~pKa-2) to turn on the common ion effect and see if the mescaline salt crashes when cold.

-
Members of the previous forum can retrieve their temporary password here, (login and check your PM).
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
Mescaline sulfate Manske?
- Thread starter Loveall
- Start date
Migrated topic.
Interesting idea, I wouldn't have thought of that. I suspect it wont work, though. The mescaline base solution will either be near saturation or quite dilute. If near saturation then the ionic effect of concentrated sulfate will lower mescaline base solubility, if very dilute you may go beyond what a common ion effect could push out in reasonable yield.
One thing I have not seen addressed in chemical literature, will a common ion effect work in an analogous way in a hydroethanolic or a nearly anhydrous solvent system? So, for example, gunk could be pushed out by bringing it to 60% alcohol and then a salt could be added to over half saturation in that solvent mix to push out the mescaline salt. Or it could be applied to harmalas.
Anyway, here is a short summary of my lab notes playing with trying to salt mescaline salts from refined solution and tea:
First I tried magnesium sulfate as its easy to find in pharmaceutical grade, this I paired with an accessory alkaloid rich cream colored cactus alkaloid hydrochloride. That did not work. Admittedly I didn't take it to the extreme that I could have but I was dubious about it to start with, suspecting that the magnesium ion would have a salting-in effect in opposition to the salting-out effect of sulfate. An amusing anecdote is here, to recover my gram of mescaline I initially based it and used xylene. Do Not do xylene pulls on freshly precipitated magnesia! It created an emulsion so bad that in the end I had to distill off the xylene and try another solvent. :lol: Saturating with salt, pulling numerous times with isopropanol and proceeding from there did work.
Next I used the same speedy, not very visual, clean cactus hydrochloride with ammonium sulfate [AS] as the salting agent. To a 2% solution of the hydrochloride I added 20g of clean AS with heat, cooled to ~0° overnight and got nothing. I reheated the solution, added another 10g of AS, cooled to ~0° overnight and got a nice crop of crystals which I isolated by vacuum filtration. To the supernatant I then added another 10g of AS, cooled to ~0° overnight and got nothing beyond a few cute crystals that would amount to no more than 1mg. The obtained needle-like crystals were recrystallized in distilled water and after being cooled to ~0° overnight I got a reasonable yield of pearlescent flake crystals. This material was assayed at 800mg in a 57 kg experienced subject and was found to be stronger, less speedy, and more visual than the starting material. The increase of potency in mescaline-like effect was in reasonable proportion to loss in gram quantity. Unexplored side note: the AS containing supernatant byproduct containing the cactus accessory alkaloid was placed at 4° for eventual isolation of that alkaloid, after some months a white solid formed in the jar that looks nothing like AS crystallized mescaline.
and was found to be stronger, less speedy, and more visual than the starting material. The increase of potency in mescaline-like effect was in reasonable proportion to loss in gram quantity. Unexplored side note: the AS containing supernatant byproduct containing the cactus accessory alkaloid was placed at 4° for eventual isolation of that alkaloid, after some months a white solid formed in the jar that looks nothing like AS crystallized mescaline.
Next I produced a tea of the european predominant scopulicola hybrid. I typically get 0,5% yield of alkaloid hydrochloride from this clone and the product is of good potency, nicely visual, and not overly speedy. Short summary is I got nothing, but here are my raw lab notes:
This is where I get confused, because my next trial makes me question the assumption that the active alkaloid we chase after is always mescaline :surprised
In this trial I extracted 200g of scop by standard method no the point of a combined xylene solution of alkaloid base. I titrated out the alkaloid to yield a pH 7 solution of alkaloid acetate. My plan was to try salting half of this directly with AS and, if it worked, adding more acid to the other half to ensure it still worked if people overshot the vinegar when pulling the alkaloid from xylene, as most people would.
I salted the first half. I got over a gram of crystal product that was clearly not AS. I tried to recrystallize in distilled water using the same parameters as before. I got absolutely no yield :shock: This is good, visual, cactus that makes me trip balls. I got a sulfate alkaloid isolate in a yield that would suggest high process efficacy, and it did not have the solubility profile of mescaline.
This suggests A) one of my favorite hallucinogenic cacti might be active due to something other than mescaline. B) my previous tea trial was trying to extract this mystery substance and not mescaline.
A possibility also exists that it was the first extraction attempt that wasn't mescaline, this I deem unlikely because the pearlescent color and shape of the crystals matched mescaline sulfate, as did its near insolubility in 0° water.
At this point I proclaimed myself in need of a break, I ate all my mescaline and proceeded to play with harmala extraction while taking 7 gram doses of mushrooms to get my equilibrium back At some point I will at least reclaim alkaloids from all that AS filled liquid. Presumably by basing, extracting to xylene, salting to water, basing with Ca(OH)2, distilling off the ammonia, extracting with xylene, and salting with HCl.
At some point I will at least reclaim alkaloids from all that AS filled liquid. Presumably by basing, extracting to xylene, salting to water, basing with Ca(OH)2, distilling off the ammonia, extracting with xylene, and salting with HCl.
One thing I have not seen addressed in chemical literature, will a common ion effect work in an analogous way in a hydroethanolic or a nearly anhydrous solvent system? So, for example, gunk could be pushed out by bringing it to 60% alcohol and then a salt could be added to over half saturation in that solvent mix to push out the mescaline salt. Or it could be applied to harmalas.
Anyway, here is a short summary of my lab notes playing with trying to salt mescaline salts from refined solution and tea:
First I tried magnesium sulfate as its easy to find in pharmaceutical grade, this I paired with an accessory alkaloid rich cream colored cactus alkaloid hydrochloride. That did not work. Admittedly I didn't take it to the extreme that I could have but I was dubious about it to start with, suspecting that the magnesium ion would have a salting-in effect in opposition to the salting-out effect of sulfate. An amusing anecdote is here, to recover my gram of mescaline I initially based it and used xylene. Do Not do xylene pulls on freshly precipitated magnesia! It created an emulsion so bad that in the end I had to distill off the xylene and try another solvent. :lol: Saturating with salt, pulling numerous times with isopropanol and proceeding from there did work.
Next I used the same speedy, not very visual, clean cactus hydrochloride with ammonium sulfate [AS] as the salting agent. To a 2% solution of the hydrochloride I added 20g of clean AS with heat, cooled to ~0° overnight and got nothing. I reheated the solution, added another 10g of AS, cooled to ~0° overnight and got a nice crop of crystals which I isolated by vacuum filtration. To the supernatant I then added another 10g of AS, cooled to ~0° overnight and got nothing beyond a few cute crystals that would amount to no more than 1mg. The obtained needle-like crystals were recrystallized in distilled water and after being cooled to ~0° overnight I got a reasonable yield of pearlescent flake crystals. This material was assayed at 800mg in a 57 kg experienced subject
and was found to be stronger, less speedy, and more visual than the starting material. The increase of potency in mescaline-like effect was in reasonable proportion to loss in gram quantity. Unexplored side note: the AS containing supernatant byproduct containing the cactus accessory alkaloid was placed at 4° for eventual isolation of that alkaloid, after some months a white solid formed in the jar that looks nothing like AS crystallized mescaline.Next I produced a tea of the european predominant scopulicola hybrid. I typically get 0,5% yield of alkaloid hydrochloride from this clone and the product is of good potency, nicely visual, and not overly speedy. Short summary is I got nothing, but here are my raw lab notes:
100g scop chips brewed 4X with a little vinegar on the first two, the liquids combined and concentrated to 225ml, settled at 4° and decanted twice yielding a slightly viscous fluid extract.
Liquid heated and 90g (NH4)2SO4 was mixed in well. A jelly-like blob of brown formed, d. <liquid. Mixture heated to near boiling to help in separation and mixture was slowly cooled to ~0°.
• Black pectinous solids collected from surface and beaker sides, weighed 14,45g, reserved at 4°.
• Thin brown liquid isolated, heated to 85° with stirring, set to slowly cool. Cooled to ~0°
• Crystals and associated thin layer of brown solids all dissolved in 20ml very hot dH2O. Cooled to ~0°
The chilled supernatant produced a few more blocky crystals that look like (NH4)2SO4.
The previous crystals [again looking like (NH4)2SO4] did not recrystallize at ~0°
To see if the pectin fraction entrained the alkaloid sulfates dH2O was added to black solids to a volume of 50ml and stirred to dissolution at at ~70°, cooled, and kept at ~0° overnight. Nothing.
The supernatant formed a layer of apparent (NH4)2SO4 crystals after several days at 4°, crystals dissolved in 20ml 80° dH2O, cooled to ~0° and held there overnight. A single (NH4)2SO4 crystal formed.
• I think this trial, as first conceived, is a fail. Retaining material for eventual reclamation.
• One explanation for a fail would be pectin holding on to the alkaloid, this could be addressed by pretreatment with pectic enzyme or acidification to pH < 2,5, the latter might fail via M-bisulfate and the former might fail by pectin monomers and oliogmers similarly salting-in mescaline
• Another possibility is that the mescaline concentration was simply too low.
Liquid heated and 90g (NH4)2SO4 was mixed in well. A jelly-like blob of brown formed, d. <liquid. Mixture heated to near boiling to help in separation and mixture was slowly cooled to ~0°.
• Black pectinous solids collected from surface and beaker sides, weighed 14,45g, reserved at 4°.
• Thin brown liquid isolated, heated to 85° with stirring, set to slowly cool. Cooled to ~0°
• Crystals and associated thin layer of brown solids all dissolved in 20ml very hot dH2O. Cooled to ~0°
The chilled supernatant produced a few more blocky crystals that look like (NH4)2SO4.
The previous crystals [again looking like (NH4)2SO4] did not recrystallize at ~0°
To see if the pectin fraction entrained the alkaloid sulfates dH2O was added to black solids to a volume of 50ml and stirred to dissolution at at ~70°, cooled, and kept at ~0° overnight. Nothing.
The supernatant formed a layer of apparent (NH4)2SO4 crystals after several days at 4°, crystals dissolved in 20ml 80° dH2O, cooled to ~0° and held there overnight. A single (NH4)2SO4 crystal formed.
• I think this trial, as first conceived, is a fail. Retaining material for eventual reclamation.
• One explanation for a fail would be pectin holding on to the alkaloid, this could be addressed by pretreatment with pectic enzyme or acidification to pH < 2,5, the latter might fail via M-bisulfate and the former might fail by pectin monomers and oliogmers similarly salting-in mescaline
• Another possibility is that the mescaline concentration was simply too low.
This is where I get confused, because my next trial makes me question the assumption that the active alkaloid we chase after is always mescaline :surprised
In this trial I extracted 200g of scop by standard method no the point of a combined xylene solution of alkaloid base. I titrated out the alkaloid to yield a pH 7 solution of alkaloid acetate. My plan was to try salting half of this directly with AS and, if it worked, adding more acid to the other half to ensure it still worked if people overshot the vinegar when pulling the alkaloid from xylene, as most people would.
I salted the first half. I got over a gram of crystal product that was clearly not AS. I tried to recrystallize in distilled water using the same parameters as before. I got absolutely no yield :shock: This is good, visual, cactus that makes me trip balls. I got a sulfate alkaloid isolate in a yield that would suggest high process efficacy, and it did not have the solubility profile of mescaline.
This suggests A) one of my favorite hallucinogenic cacti might be active due to something other than mescaline. B) my previous tea trial was trying to extract this mystery substance and not mescaline.
A possibility also exists that it was the first extraction attempt that wasn't mescaline, this I deem unlikely because the pearlescent color and shape of the crystals matched mescaline sulfate, as did its near insolubility in 0° water.
At this point I proclaimed myself in need of a break, I ate all my mescaline and proceeded to play with harmala extraction while taking 7 gram doses of mushrooms to get my equilibrium back
At some point I will at least reclaim alkaloids from all that AS filled liquid. Presumably by basing, extracting to xylene, salting to water, basing with Ca(OH)2, distilling off the ammonia, extracting with xylene, and salting with HCl.Practical notes on obtaining usable ammonium sulfate:
Many would not want to pay the cost of buying a kilo of technical grade ammonium sulfate by mail. They may notice that ammonium sulfate is sold as a common cheap fertilizer. This grade of ammonium sulfate is produced as a byproduct of steel manufacture, as such it contains traces of metal scale and a very fine dispersion of a oily lubricant of some sort, you should not use fertilizer AS without at least some attempt at purification.
The most extremely basic purification step would be to dissolve the crystals in hot water and cook this solution well so the lubricant mist will melt and congeal together into a brown scum that can be filtered out with the traces of metal scale. Run the solution through the same paper filter repeatedly until it comes out clear.
Further purification can be done by heating a concentrated solution and adding alcohol before cooling to ~0°. The alcohol will increase solubility of lubricant residues while crashing out AS as fine crystals.
Using a concentrated AS solution and ethyl alcohol, for every 100ml solution adding 50ml ethanol gave me 61% yield, adding 100ml ethanol gave me 88% yield, adding 225ml ethanol gave me 97% yield. Initially the AS salts out the ethanol. Vigorous stirring at ~0° is essential for proper crystal formation, my yields represent the use of a magnetic stirrer and an ice bath. Even at ice cold temperature the salting out effect is replaced with crystallization only with stirring.
Obviously this dual solvent recrystallization is most practical if you can safely reclaim the alcohol by distillation. This wont be possible for everyone which is why I mentioned the progressive yield with alcohol volume.
Many would not want to pay the cost of buying a kilo of technical grade ammonium sulfate by mail. They may notice that ammonium sulfate is sold as a common cheap fertilizer. This grade of ammonium sulfate is produced as a byproduct of steel manufacture, as such it contains traces of metal scale and a very fine dispersion of a oily lubricant of some sort, you should not use fertilizer AS without at least some attempt at purification.
The most extremely basic purification step would be to dissolve the crystals in hot water and cook this solution well so the lubricant mist will melt and congeal together into a brown scum that can be filtered out with the traces of metal scale. Run the solution through the same paper filter repeatedly until it comes out clear.
Further purification can be done by heating a concentrated solution and adding alcohol before cooling to ~0°. The alcohol will increase solubility of lubricant residues while crashing out AS as fine crystals.
Using a concentrated AS solution and ethyl alcohol, for every 100ml solution adding 50ml ethanol gave me 61% yield, adding 100ml ethanol gave me 88% yield, adding 225ml ethanol gave me 97% yield. Initially the AS salts out the ethanol. Vigorous stirring at ~0° is essential for proper crystal formation, my yields represent the use of a magnetic stirrer and an ice bath. Even at ice cold temperature the salting out effect is replaced with crystallization only with stirring.
Obviously this dual solvent recrystallization is most practical if you can safely reclaim the alcohol by distillation. This wont be possible for everyone which is why I mentioned the progressive yield with alcohol volume.
Elrik, this is great. Thank you!
Reading through your notes the first thing that jusmps to mind is that a simple water extraction won't work. It will be too gunky to concentrate to 1% without making a gel. Instead, we would want to treat it with acetone or IPA to precipitate the junk, or perhaps start with an ethanol extract. Secondly, it appears that ammonium sulfate works better than magnesium sulfate?
The water/miscible solvent/salt system is very interesting. We have some discussion of that in this thread
That being said, I have questions,
1) When you tried magnesium sulfate, you had ~1g of alkaloids, how much volume of water did you use? How much salt did you add?
2) Your next test is fascinating (2% alkaloid hydrochlorides in water with ammonium sulfate added). How much water did you add? How much of the starting material do you think was mescaline? Trying to understand h, and c (estimated mescaline and added salt concentration) so I can reproduce.
Thanks!
Reading through your notes the first thing that jusmps to mind is that a simple water extraction won't work. It will be too gunky to concentrate to 1% without making a gel. Instead, we would want to treat it with acetone or IPA to precipitate the junk, or perhaps start with an ethanol extract. Secondly, it appears that ammonium sulfate works better than magnesium sulfate?
The water/miscible solvent/salt system is very interesting. We have some discussion of that in this thread
That being said, I have questions,
1) When you tried magnesium sulfate, you had ~1g of alkaloids, how much volume of water did you use? How much salt did you add?
2) Your next test is fascinating (2% alkaloid hydrochlorides in water with ammonium sulfate added). How much water did you add? How much of the starting material do you think was mescaline? Trying to understand h, and c (estimated mescaline and added salt concentration) so I can reproduce.
Thanks!
With the magnesium sulfate I started with 100ml of a 1% solution of the clean alkaloid hydrochloride in distilled water and dissolved in 20g of MgSO4•7H2O and cooled to 4° for a day. Even after seeding with an evaporated drop once it was cold it produced no crystal. I did not try a higher salt concentration due to magnesium sulfate solubility constraints.
It should also be kept in mind that ammonium sulfate is twice as concentrated a source of sulfate ions, compared to magnesium sulfate, and even ammonium sulfate gave no crystallization at 20g/100ml solution. Lithium sulfate would probably be great, but who has kilos of that? :lol:
On the successful fractionation I mentioned it was hard to guess exactly how much of the alkaloid was mescaline, since the accessory alkaloid was active at least as a stimulant, but I was guessing 1/3 to 1/2 mescaline based on the visuals and mental effects.
I actually did two runs using ammonium sulfate. On the first it was 1,00 grams of alkaloid hydrochloride in 100ml dH2O. In this I tried a total of 10, 20, 40, and 45 grams AS. All obtainable product was produced at the 40g level with the product weighing 1,13g vacuum filtered and dried. This was then recrystallized from 15ml water cooling from 80° to ~0° to yield 0,48g of the pearlescent flakes [this was already crystallizing at 20°] and, assuming the product was a sulfate dihydrate, that means I got 42,5% theoretical yield. I'm assuming most of the accessory alkaloid was salted out but stayed in solution upon recrystallization.
The second run was to ensure I had enough material for a valid test, since I rarely consume low doses In it I put 2,14g •HCl into 100ml dH2O and crashed it out with 30g AS to yield 1,16g [54% theoretical yield] after recrystallization.
Raw lab notes:
It should also be kept in mind that ammonium sulfate is twice as concentrated a source of sulfate ions, compared to magnesium sulfate, and even ammonium sulfate gave no crystallization at 20g/100ml solution. Lithium sulfate would probably be great, but who has kilos of that? :lol:
On the successful fractionation I mentioned it was hard to guess exactly how much of the alkaloid was mescaline, since the accessory alkaloid was active at least as a stimulant, but I was guessing 1/3 to 1/2 mescaline based on the visuals and mental effects.
I actually did two runs using ammonium sulfate. On the first it was 1,00 grams of alkaloid hydrochloride in 100ml dH2O. In this I tried a total of 10, 20, 40, and 45 grams AS. All obtainable product was produced at the 40g level with the product weighing 1,13g vacuum filtered and dried. This was then recrystallized from 15ml water cooling from 80° to ~0° to yield 0,48g of the pearlescent flakes [this was already crystallizing at 20°] and, assuming the product was a sulfate dihydrate, that means I got 42,5% theoretical yield. I'm assuming most of the accessory alkaloid was salted out but stayed in solution upon recrystallization.
The second run was to ensure I had enough material for a valid test, since I rarely consume low doses
In it I put 2,14g •HCl into 100ml dH2O and crashed it out with 30g AS to yield 1,16g [54% theoretical yield] after recrystallization.Raw lab notes:
171130 Refined alkaloid + Magnesium sulfate:
20,00g of MgSO4•7H2O was dissolved in 100 ml distilled water and cooled to 6°C. A seed crystal was added and the solution was returned to the cold. As expected, the seed crystal dissolved.
The solution was heated to 58°C and 1,00g of off-white acetone washed KG X SS02 alkaloid hydrochloride was dissolved in. A single drop was drawn out to evaporate and the beaker was covered and placed in a cold water bath in the refrigerator.
When the solution had cooled to 4°C the evaporated drop was stirred in to the solution, and the beaker was returned to the refrigerator and left there overnight. No crystals formed.
100 ml of sat. NaCl added and the beaker was returned to the fridge. No crystals formed.
Is the magnesium preventing crystallization by the formation of a coordination complex?
If so, another sulfate should work.
===
§§§
===
Refined alkaloid + Ammonium sulfate:
1,00g cream colored KG X SS02 alkaloid•HCl dissolved in sufficient dH2O to make 100ml.
Solution heated to 60° and 10,00g of (NH4)2SO4 stirred in. Solution cooled. Nothing.
Another 10,00g stirred in. Solution cooled. Nothing.
20,00g more stirred in to give immediate crystallization in warm solution. Solution cooled to 0° and vacuum filtered after some hours.
Another 5,00g stirred in. Solution cooled to 0°. Trivially small quantity of crystals.
Dry 1,13g of collected crystals [90% yield] dissolved in 15ml near boiling water. Cooling to room temp initiated crystal formation. Cooling to 4° created more. Solution held at 0° overnight and vacuum filtered yielding 0,48g [42,5% yield] of bitter crystals that are clearly not (NH4)2SO4.
• Assuming the crude 1,13g of product was not half (NH4)2SO4 it is possible I have
separated mescaline from the mystery alkaloid in the KG X SS02 alkaloid•HCl, peers support this
Save both fractions and test both, by then a scop•HCl test result or two should be in.
Supernatant warmed, mixed with an equal volume of ethanol, and cooled in an ice bath. <0,5mg of needle-like crystals formed.
Solution warmed, mixed with another volume of ethanol, and cooled in an ice bath. <1mg of needle-like crystals formed. Alcoholic supernatant reserved at 4°
===
§§§
===
Refined alkaloid + Ammonium sulfate #2:
2,14g cream colored KG X SS02 alkaloid•HCl dissolved in sufficient dH2O to make 100ml.
Solution heated to 80° and 30,00g of (NH4)2SO4 stirred in. Solution filtered and cooled, with crystallization commencing at ~60°. Cooling continued to ~0°, temp maintained several hours.
Mixture vacuum filtered, supernatant reheated to 85° and another 10,00g of (NH4)2SO4 stirred in. Solution filtered, cooled, and held at ~0° for 13 hours. No crystals.
Crystals dissolved in 30ml 85° dH2O, filtered, and crystallized holding at ~0° for 13 hours. Recrystallized product obtained by vacuum filtration dried to 1,16g [54% yield]. Recrystallization supernatant combined with that of the previous run and reserved for accessory alkaloid reclamation.
20,00g of MgSO4•7H2O was dissolved in 100 ml distilled water and cooled to 6°C. A seed crystal was added and the solution was returned to the cold. As expected, the seed crystal dissolved.
The solution was heated to 58°C and 1,00g of off-white acetone washed KG X SS02 alkaloid hydrochloride was dissolved in. A single drop was drawn out to evaporate and the beaker was covered and placed in a cold water bath in the refrigerator.
When the solution had cooled to 4°C the evaporated drop was stirred in to the solution, and the beaker was returned to the refrigerator and left there overnight. No crystals formed.
100 ml of sat. NaCl added and the beaker was returned to the fridge. No crystals formed.
Is the magnesium preventing crystallization by the formation of a coordination complex?
If so, another sulfate should work.
===
§§§
===
Refined alkaloid + Ammonium sulfate:
1,00g cream colored KG X SS02 alkaloid•HCl dissolved in sufficient dH2O to make 100ml.
Solution heated to 60° and 10,00g of (NH4)2SO4 stirred in. Solution cooled. Nothing.
Another 10,00g stirred in. Solution cooled. Nothing.
20,00g more stirred in to give immediate crystallization in warm solution. Solution cooled to 0° and vacuum filtered after some hours.
Another 5,00g stirred in. Solution cooled to 0°. Trivially small quantity of crystals.
Dry 1,13g of collected crystals [90% yield] dissolved in 15ml near boiling water. Cooling to room temp initiated crystal formation. Cooling to 4° created more. Solution held at 0° overnight and vacuum filtered yielding 0,48g [42,5% yield] of bitter crystals that are clearly not (NH4)2SO4.
• Assuming the crude 1,13g of product was not half (NH4)2SO4 it is possible I have
separated mescaline from the mystery alkaloid in the KG X SS02 alkaloid•HCl, peers support this
Save both fractions and test both, by then a scop•HCl test result or two should be in.
Supernatant warmed, mixed with an equal volume of ethanol, and cooled in an ice bath. <0,5mg of needle-like crystals formed.
Solution warmed, mixed with another volume of ethanol, and cooled in an ice bath. <1mg of needle-like crystals formed. Alcoholic supernatant reserved at 4°
===
§§§
===
Refined alkaloid + Ammonium sulfate #2:
2,14g cream colored KG X SS02 alkaloid•HCl dissolved in sufficient dH2O to make 100ml.
Solution heated to 80° and 30,00g of (NH4)2SO4 stirred in. Solution filtered and cooled, with crystallization commencing at ~60°. Cooling continued to ~0°, temp maintained several hours.
Mixture vacuum filtered, supernatant reheated to 85° and another 10,00g of (NH4)2SO4 stirred in. Solution filtered, cooled, and held at ~0° for 13 hours. No crystals.
Crystals dissolved in 30ml 85° dH2O, filtered, and crystallized holding at ~0° for 13 hours. Recrystallized product obtained by vacuum filtration dried to 1,16g [54% yield]. Recrystallization supernatant combined with that of the previous run and reserved for accessory alkaloid reclamation.
Thank you so much, Elrik this data is great 
A couple comments:
1) Looks like you used MgSO4°7H2O (based on the 2x sulfate for AS comment). I think your results can be explained by looking at the concentration of SO4-- ions. While it could be true, there is no need to invoke Mg++ effects I think: performance of both salts matched at the common SO4-- concentration tested of only 0.74M: at that concentration neither salt crashed crystals.
2) AS started to crash mescaline after 40g (~2.2M, linear extrapolation from 74.4g AS in 100g of water = 4.1M). Incidentally, this means the solution grew to a volume of ~140ml.
3) In summary, in the succesfull manske, h~0.014M and c ~ 2.2M. This is very helpful, I'll try to reproduce. By they way, using s = 0.02 in the previous equations would have given r = 86% with this setup (however, it also predicts significant crashing at the lower c of 1.1M, but that didn't happen , so it doesn't seem the equations are that representative.
, so it doesn't seem the equations are that representative.
A couple comments:
1) Looks like you used MgSO4°7H2O (based on the 2x sulfate for AS comment). I think your results can be explained by looking at the concentration of SO4-- ions. While it could be true, there is no need to invoke Mg++ effects I think: performance of both salts matched at the common SO4-- concentration tested of only 0.74M: at that concentration neither salt crashed crystals.
2) AS started to crash mescaline after 40g (~2.2M, linear extrapolation from 74.4g AS in 100g of water = 4.1M). Incidentally, this means the solution grew to a volume of ~140ml.
3) In summary, in the succesfull manske, h~0.014M and c ~ 2.2M. This is very helpful, I'll try to reproduce. By they way, using s = 0.02 in the previous equations would have given r = 86% with this setup (however, it also predicts significant crashing at the lower c of 1.1M, but that didn't happen
, so it doesn't seem the equations are that representative.Yes, when the magnesium test failed I wasn't sure if it was the magnesium interference or sulfate concentration issue. Since AS worked at 30g but not 20g that means for an equivalent sulfate ion situation I would have to use over 40g, possibly almost 60g of magnesium sulfate heptahydrate while cooling to 0°, which clearly would not work due to solubility constraints.
Some day I really have to figure out why it didn't work with a refined scop acetate solution. The most likely scenarios are either acetate somehow blocks the common ion effect of sulfate, though I have no idea how it would, or the active constituent of my scop clone is not mescaline, which would be remarkable to say the least.
Soon I should reclaim those grams of scop alkaloid from the AS mess and try again, this time with scop hydrochloride. That way I don't dump another quarter kilo of my beloved scop chips into making more ammonium sulfate-mescaline solution mess :lol:
Do you see any better way of separating cactus alkaloids and AS than my plan of basing, extracting to xylene, salting to water, basing with Ca(OH)2, distilling off the ammonia, extracting with xylene, and salting with HCl? I think that should work, and a trace of ammonium chloride wouldn't be toxic if it was less than 100% effective, but the route seems kind of circuitous.
Some day I really have to figure out why it didn't work with a refined scop acetate solution. The most likely scenarios are either acetate somehow blocks the common ion effect of sulfate, though I have no idea how it would, or the active constituent of my scop clone is not mescaline, which would be remarkable to say the least.
Soon I should reclaim those grams of scop alkaloid from the AS mess and try again, this time with scop hydrochloride. That way I don't dump another quarter kilo of my beloved scop chips into making more ammonium sulfate-mescaline solution mess :lol:
Do you see any better way of separating cactus alkaloids and AS than my plan of basing, extracting to xylene, salting to water, basing with Ca(OH)2, distilling off the ammonia, extracting with xylene, and salting with HCl? I think that should work, and a trace of ammonium chloride wouldn't be toxic if it was less than 100% effective, but the route seems kind of circuitous.
Elrik said:Yes, when the magnesium test failed I wasn't sure if it was the magnesium interference or sulfate concentration issue. Since AS worked at 30g but not 20g that means for an equivalent sulfate ion situation I would have to use over 40g, possibly almost 60g of magnesium sulfate heptahydrate while cooling to 0°, which clearly would not work due to solubility constraints.
Some day I really have to figure out why it didn't work with a refined scop acetate solution. The most likely scenarios are either acetate somehow blocks the common ion effect of sulfate, though I have no idea how it would, or the active constituent of my scop clone is not mescaline, which would be remarkable to say the least.
Soon I should reclaim those grams of scop alkaloid from the AS mess and try again, this time with scop hydrochloride. That way I don't dump another quarter kilo of my beloved scop chips into making more ammonium sulfate-mescaline solution mess :lol:
Do you see any better way of separating cactus alkaloids and AS than my plan of basing, extracting to xylene, salting to water, basing with Ca(OH)2, distilling off the ammonia, extracting with xylene, and salting with HCl? I think that should work, and a trace of ammonium chloride wouldn't be toxic if it was less than 100% effective, but the route seems kind of circuitous.
Dry magnesium sulfate dissolves up to 26g/100mg at 0C, so maybe there is a small process corner where it may work (?). I think we would want 2% or more for alkaloid concentration.
Separating SA: I would guess you can outgas ammonia right after the first basing (or am I missing something?). Not sure what the ammonia partition coefficient into xylene is, some may be removed there too (if any remains). I agree ammonium chloride traces would not be toxic, it is used in food (as you may know) such as salty licorice.
I didn't think directly outgassing the ammonia after the initial basing was a viable plan due to the sheer amount of AS. Either I would have to dump in a ton of lye to convert it all to ammonia and make my house smell like a meth lab from a kilometre away :lol: or I would be left with a solution of alkaloid base and no other base in concentrated AS, probably with some alkaloid carbonate forming. I'm not sure how mescaline, or especially more basic accessory alkaloids, would behave in a water/xylene partition with a large quantity of AS and no available inorganic base.
From the data I've seen, ammonias solubility in xylene is not high and it would largely prefer water in a partition. So that first xylene partition should remove all AS and the larger portion of ammonia while recovering nearly all alkaloid, assuming I did 5 pulls.
I'm confident my reclamation route will work, provided Ca(OH)2 powder doesn't create a nasty xylene-water emulsion, it's just time consuming and so I hoped I was missing something blatantly obvious. This is the main reason I tried magnesium sulfate before ammonium sulfate
From the data I've seen, ammonias solubility in xylene is not high and it would largely prefer water in a partition. So that first xylene partition should remove all AS and the larger portion of ammonia while recovering nearly all alkaloid, assuming I did 5 pulls.
I'm confident my reclamation route will work, provided Ca(OH)2 powder doesn't create a nasty xylene-water emulsion, it's just time consuming and so I hoped I was missing something blatantly obvious. This is the main reason I tried magnesium sulfate before ammonium sulfate
Did more tests on cacti. 75% everclear resin (very bitter) from 50g of cacti powder was dried and re-dissolved in water (35ml), decanted/filtered, resulting in a dark and opaque solution.
Ammonium sulfate was added to a 5ml sample of this. Gunky precipitation started at ~15% salt and was massive at 25%, at which point the solution beame a clear amber. At 35% ammonium sulfate a little bit more gunk precipitated and not much after that. I decanted the solution at several ammonium sulfate concentrations while warm and put it in the fridge hoping to see mescaline sulfate needles form. That did not happen, instead gunk precipitated.
All the crashed gunk from the ammonium sulfate crashes was bitter. It seems to me that both mescaline and plant stuff crashed together and I wasn't able to separate them.
I only used the bitter taste as a way to track where the mescaline was, so I could be wrong, but those are my rough observations at this time.
Ammonium sulfate was added to a 5ml sample of this. Gunky precipitation started at ~15% salt and was massive at 25%, at which point the solution beame a clear amber. At 35% ammonium sulfate a little bit more gunk precipitated and not much after that. I decanted the solution at several ammonium sulfate concentrations while warm and put it in the fridge hoping to see mescaline sulfate needles form. That did not happen, instead gunk precipitated.
All the crashed gunk from the ammonium sulfate crashes was bitter. It seems to me that both mescaline and plant stuff crashed together and I wasn't able to separate them.
I only used the bitter taste as a way to track where the mescaline was, so I could be wrong, but those are my rough observations at this time.
I suspect the gunk is largely comprised of pectins and the hyaluronic acid-like compounds known to be in cacti, compounds which could entrap alkaloids due to their acidity the same way tannin does in aya.
Dropping the pH to 2 or less would be expected to free the alkaloids from these compounds, and then it would just be a question of if mescaline sulfate would crystallize at 0° when it could be acidic enough to encourage bisulfate formation. Protonating hyaluronic acid-like compounds also makes them less mucilaginous.
Dropping the pH to 2 or less would be expected to free the alkaloids from these compounds, and then it would just be a question of if mescaline sulfate would crystallize at 0° when it could be acidic enough to encourage bisulfate formation. Protonating hyaluronic acid-like compounds also makes them less mucilaginous.
Elrik said:I suspect the gunk is largely comprised of pectins and the hyaluronic acid-like compounds known to be in cacti, compounds which could entrap alkaloids due to their acidity the same way tannin does in aya.
Dropping the pH to 2 or less would be expected to free the alkaloids from these compounds, and then it would just be a question of if mescaline sulfate would crystallize at 0° when it could be acidic enough to encourage bisulfate formation. Protonating hyaluronic acid-like compounds also makes them less mucilaginous.
That is very interesting. I did not know about hyaluronic acid-like compounds. It looks like it forms a large matrix that entraps material?
Once I did an IPA pull on spent cacti (used for lime/limo tek). I was acidifying the pulled IPA with sulfuric acid, but I overshot to pH 1.9, never seeing crystals. Then I raised the pH and needle like xtals formed.
However, would a strong alkaline setup not break down hyaluronic acid-like stuff? Before the IPA pull, the cacti powder was treated with NaOH. Did the extra acid really do something to the plant material that helped with xtal formation?
Strong base [and, to a lesser extent, strong acids] will hydrolyze pectins and polysaccharides, but it often would only be partial unless the pH approached very high levels. I use lye to base my thick cactus tea to quite a strong pH and it still increases viscosity by deprotonating the acid groups while not hydrolyzing the gunk enough to counter the effect. I always have to make my tea thinner than the maximum viscosity I can pull to accommodate the base I'll be adding.
Over in the nook archives I've read the exploits of cactus chemists who based to lye saturation to make the solution thin but I've never approached that level of danger/waste. :lol:
Over in the nook archives I've read the exploits of cactus chemists who based to lye saturation to make the solution thin but I've never approached that level of danger/waste. :lol:
Regarding proteins, maybe there is a way to separate them from the alkaloids. See attached paper where they claim to separate the two with a very simple method.
To retrieve the alkaloids:
To retrieve the alkaloids:
That apparently leaves the proteins in the filtered slurry and the mescaline in the chilled acetone. We could try chilled ~85% aqueous acetone extraction before manske for protein/alkaloid separation.Attached Paper said:The plants were removed from the greenhouse in pairs and sliced and blended with 5 vol. of chilled acetone (-15C), at high speed, for 1 min in a Waring blender."
Attachments
:lol: I actually did consider that method once, until I thought it through and realized the large amount of acetone involved and the subsequent recovery by distillation of all that.
And it would also crash out any acidic polysaccharides, so acid would have to be added to free the mescaline from that first, and then you'd have the problem of aldol condensations by stewing acetone [and acetone+alkaloid] with acid.
And it would also crash out any acidic polysaccharides, so acid would have to be added to free the mescaline from that first, and then you'd have the problem of aldol condensations by stewing acetone [and acetone+alkaloid] with acid.
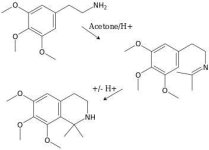
downwardsfromzero
Boundary condition
I also wonder about Pictet-Spengler cyclisation of the acetone-alkaloid iminium adduct to produce an isoquinoline (~1,1-dimethylanhalinine). While this reaction would seem to be possible, perhaps it would be sufficiently slow to be of lesser concern in yield losses. TLC would (or should possibly) give some indication as to whether this side-reaction occurs. It's more than a little disfavoured sterically, I suppose.Elrik said::lol: I actually did consider that method once, until I thought it through and realized the large amount of acetone involved and the subsequent recovery by distillation of all that.
And it would also crash out any acidic polysaccharides, so acid would have to be added to free the mescaline from that first, and then you'd have the problem of aldol condensations by stewing acetone [and acetone+alkaloid] with acid.
I also wonder to what degree aldose sugars might eat into mescaline yields with prolonged stewing. And then we have peyoruvic acid as an example of a related condensation product, in Loph. w., at least, where the alkaloid production has crossed paths with a major metabolic intermediate. Just swap one of the methyls at the 1-position for a carboxylic acid group in the putative acetone/mesc reaction scheme attached.
Attachments
Similar threads
- Replies
- 3
- Views
- 534
- Replies
- 1
- Views
- 635
- Replies
- 46
- Views
- 4K