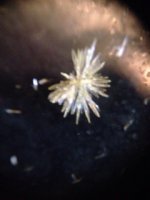
psilocybin
Rising Star
Any questions or problems you may run into during an extraction can be asked to get other members input in this thread.

No. Citric acid is not volatile. Browse the forum a bit more, there is plenty of info here so you won't have to re-invent the wheel.can you use citric acid saturated acetone with a Soxhlet extractor?

")
")
In order, to the best of my limited knowledge :JefFlux said:A set of theoretical questions (and apologies if these are somewhere in the FAQ);
Does the length of time that solvent sits on base tea (between rolls) increase crossover of the molecule into the solvent?
Does the solvent need to 'make contact' with as much base tea as possible - or is this mostly a chemical transference from polar to non-polar?
Is there an optimum method of agitation? Stirring seems to work and dos not run the risk of emulsification but I ma wondering if this is sufficient
thanks in advance,
Flux