Summary: I purified a larger mass of mesembrine-dominant extract by flash chromatography, yielding a pale yellow oil. I attempted to crystallize as the fumarate, succinate, oxalate, and malate. The fumarate precipitates from acetone within minutes. The malate doesn't, but recrystallizes from isopropanol. Both form clear, colorless, odorless solutions in water. The succinate was always an oil, and the oxalate was a mess.
Purification by Flash Chromatography
I started with 1017 mg crude alkaloids, obtained by CIELO-style extraction into ethyl acetate, salting into water, and extraction back to the base. I used material from a plant that naturally produced mostly mesembrine, and I did not attempt to separate mesembrine from mesembrenone. I therefore used a lot less silica than before, 5 g. I loaded wet this time, and eluted with 7:3:0.1 EtOAc:heptane:NH3(aq) as before. I collected fractions of 12 mL, and analyzed by TLC as before.
Fraction 1 was 51 mg and strongly odorous, but did not contain mesembrine alkaloids. I pooled fractions 2 and 3 and evaporated to obtain 491 mg of a pale yellow oil.

I pooled fractions 4-7, obtaining 150 mg. No subsequent fractions contained mesembrine alkaloids. This is inconsistent with my previous run, where the alkaloids ran much slower even adjusting for the longer column and smaller fractions. I can't explain the big difference, and I suspect a mistake in my previous run. The results this time seem more consistent with the TLC. The color stayed almost entirely on the column as before.
I'm happy with this separation. A plug of silica in a Buchner funnel might also work, though to remove the odor and taste it would be necessary to load and elute separately and discard the first fraction, rather than just filtering.
Crystallization of Salts
The best reference on mesembrine salts appears to be UK Patent Application GB 2619907. They screened multiple solvents, including MEK but not acetone. They report twelve counterions, including positive results on the succinate and fumarate and negative on the citrate, benzoate and maleate. Their most stable salt was the besylate (benzenesulfonate), which I was surprised to learn is used in several other approved drugs. I still didn't want to mess with it though. The hydrochloride looks pretty good too, but I didn't want to bother preparing water-free organic HCl solutions.
I screened the fumarate, succinate, oxalate, and malate. I was reasonably careful to exclude water. I dried my acetone over sodium sulfate and distilled it a few days before use. When evaporating fractions I heated to 90 C under moderate (~5 kPa) vacuum. All containers were capped when not actively working.
To each of four weighed 1.5 mL vials I added 1.1 equivalents of the acid and sonicated to dissolve in acetone. I then added a solution of the purified mesembrine (fractions 2-3) in acetone, around 200 mg/mL. For the fumaric acid I used 1.2 mL acetone to dissolve the acid and 40 mg mesembrine. For the others I used 0.75 mL acetone and 50 mg mesembrine, but the fumaric acid isn't soluble enough for that.
The fumarate crystallized within minutes. The patent application says theirs was "poorly crystalline" from MEK. I don't know if mine from acetone would make sharp peaks in the XRD, but the crystals are pretty and free-flowing. I carefully syringed off the mother liquor, dried the crystals under an air stream, and weighed them in the vial by subtraction yielding 43 mg.

None of the others salts showed crystallization after ten hours at room temperature (including the succinate, which was crystalline from their MEK). I put them all in the freezer for nine hours, after which the oxalate and malate showed an oil or resin that quickly redissolved upon warming.
I tried crystallizing the other three from ethanol but they were all too soluble, dissolving easily in 0.1 mL. I added 1 mL ethyl acetate as an antisolvent, which yielded an oil in the malate, a very fine possible solid in the oxalate that reverted to an oil after about an hour, and no change in the succinate.
I tried crystallizing from isopropanol. I sonicated the malate with 1 mL at 50 C. It wouldn't fully dissolve, but fine white crystals appeared on the bottom of the malate vial. This felt promising, so I added ethanol until it did fully dissolve (which was 90 uL), then left the vial to cool and crystallize. This yielded white crystals, though they weren't as free-flowing as the fumarate (though I also might not have dried them enough, with the higher-boiling solvent). Pure isopropanol would probably work at higher temperature and lower concentration, or maybe ethanol/isopropanol as the solvent/antisolvent.
The oxalate also formed a possible solid in isopropanol, but one so fine it seemed hard to separate from the mother liquor. The succinate dissolved completely in 0.5 mL isopropanol and remained clear after an hour or so. I evaporated the succinate down to 0.25 mL, and saw no change after eight hours. I added 0.5 mL heptane and it oiled out. I added 0.4 mL ethanol and it redissolved after long sonication, but then it stayed dissolved on cooling. I froze it, and it oiled out.
I evaporated the solvent from the two failures, recovering a pale yellow oil in each. I redissolved all four salts in 1 mL water each. The fumarate and malate crystals each made an odorless, water clear solution. The succinate and oxalate resin made a cloudy solution, I assume from a nonpolar impurity that crystallization separated. After filtration through a syringe filter they became a clear, very pale yellow.

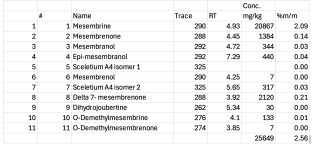
Analysis shows that the malate is 95% pure by UV area, and the fumarate is 98%. The remainder is mesembrenone, which was depleted by both the chromatography and the crystallization. Fractions 4-7 are about 80% mesembrine and 20% mesembrenone, correspondingly enriched. The early peak in the fumarate is probably the counterion. The other three counterions probably elute around the same time, but lack the double bond and thus don't absorb.

I now feel some optimism that crystallization could be possible without chromatography, since I'd chosen bad counterions for my previous experiment. I'll retry next time with the fumarate from acetone and malate from isopropanol.
Miscellaneous
@Woolmer's lab's HPLC method looks pretty typical, alkaline conditions on a C18 column. The two epimesembranol peaks still seem mysterious.
I'm quite confident that any bitter taste of
Sceletium is mostly not mesembrine, since the purified mesembrine citrate extract tastes much less bitter than crude alkaloid citrate. Extract purified by crystallization has almost no taste, just very slightly bitter and sour.
I think my residual odor and taste after chromatography but before crystallization is from my ethyl acetate, maybe some kind of higher ester impurity. I tried distilling my ethyl acetate and that might have helped somewhat, but not completely. Maybe activated carbon would help. It doesn't come off under 5 kPa vacuum; maybe higher vacuum would help. Perhaps this doesn't matter, since I'll crystallize all material for consumption going forward.
At higher dose, I think mesembrine and mesembrenone are distinguishable, with the former giving more intense but increasingly unpleasant effects while the latter plateaus. Neither seems particularly abusable to me. That said, I'm one person and even some heroin users just don't get addicted, so proceed carefully. Kanna has long history of safe use, but more purified drugs are often more abusable, as demonstrated by refined cocaine, vodka, etc.
I might next try synthesizing mesembranols by reduction of mesembrine. These are natural products but with small abundance, so more easily produced semi-synthetically. Patnala separated the epimers by prep HPLC but there must be a better way, maybe on a C18 SPE cartridge.
A startup is working to bring some mesembrine analogs to market, and PCT Application WO2025101939A1 gives the best SAR insight I've seen published. I haven't studied carefully, but I think most or all of their modifications are achievable only by total synthesis.
I've grown some of my clones outdoors. I harvested one batch this spring during vegetative growth, which I've now finally analyzed. I wasn't expecting much, since I was watering daily; but it's actually lower water content than my indoor material, and roughly as strong. This may be due to nearby
Passiflora, which competes aggressively for water and is also known to be allelopathic (and may in fact be killing the
Sceletium, since the latter is sickly now and I can't otherwise explain that). I took a later harvest when flowers were present, with similar alkaloid ratio but less than a quarter the absolute alkaloids.
I usually freeze my plant material for storage before I get around to drying it, but I recently dried some unfrozen material in a food dehydrator. The leaf cuticle seems to mostly remain intact, eventually producing a hollow shell. This makes an odd crinkling noise when it eventually crumbles, and also greatly slows drying. I got impatient and milled the damp material to break up the leaves, then resumed drying. I think freezing is probably beneficial if only to disrupt the cuticle, and results from Glyn-Woods may imply that preserves the alkaloids better too.
Handle these extracts with care, since they're active around 1 mg. Accidental exposure is unlikely to be fatal, but an overdose would at least be unpleasant and impairment while working could result in a serious accident. The crystals are easier to mishandle than the resins since they don't stick together. These extracts must always be diluted for consumption.