
All the reports I've seen suggest the same. The melting point is recorded as 32-35°C, but it's said to persist as an oil at temperatures well below this.I may be showing my unfamiliarity with mescaline again, but I think you'll get a sticky oil.
Just bear in mind, freebase mescaline is particularly noted for formation of a solid carbonate (or carbamate/bicarbonate - this is something I've been meaning to look into) by absorption of CO₂ and maybe water vapour from the atmosphere. This has the potential to throw any weigh calculations off by nearly as much as the uncertainties with the salt forms. Well, somewhere between 22 and 72 mass units on top of mescaline's 211, depending on the stoich«ometry, more if there's any additional hydration.The easiest way I've found to evaporate solvent is a water bath below the boiling point of the solvent, with air blowing on the solvent surface.
On the plus side, at least if it's not actually its own ipsocarbamate, the salt will be unstable and probably decomposes below 100°C, looking at NaHCO₃ as an analogy.
C₁₁H₁₅O₃NH₃⁺.HCO₃⁻ → C₁₁H₁₅O₃NH₂ + H₂O + CO₂
There's also a question of how hygroscopic this freebase might be, given its relatively high solubility in water. There' a certain amount of digging into the literature to be done in order to confirm whether there is any firm data regarding these points.
Investigating them practically for oneself would seem to require a few items of lab equipment, or at least the ability to creatively improvise them.
Don't be put off, however - I'm hoping that by providing some foresight it will help experiments to go more smoothly. It looks like 'simply' warming the carbonate-contaminated freebase under a controlled atmosphere and slightly reduced pressure should get rid of much of the CO₂ and moisture.
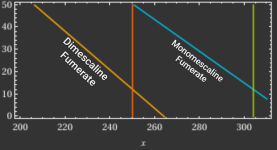
 ) and pulling with ethyl acetate, then re-salting citric acid to compare weight vs 400mg fumarate. I've never isolated freebase mescaline before I don't have a clue offhand how to do that to weigh it for comparison
) and pulling with ethyl acetate, then re-salting citric acid to compare weight vs 400mg fumarate. I've never isolated freebase mescaline before I don't have a clue offhand how to do that to weigh it for comparison  .
.
")

