I have a three-part update today:
Phalaris aquatica
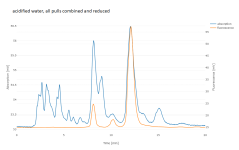
I had 273 g P. aquatica (with unknown genetics) in my freezer, so I extracted that too. I used basically the same method, in two pulls into 1411 mL water, then reduced to 50 mL and filtered. Even allowing for the greater reduction ratio, this seemed worse-behaved than the P. brachystachys, with lots of waxy stuff floating to the top while reducing (which I skimmed off), and heavy foam in the flask while filtering under vacuum. (Maybe lower temperature would extract less unwanted stuff?) The aqueous extract contained only 29 mg DMT and 3 mg 5-MeO-DMT, which didn't seem worth extracting further.
Overall, this reaffirms my plan to focus on my P. brachystachys, especially since others are working with much better P. aquatica genetics. To emphasize, my results apply only to the particular seeds I purchased, since intra-species variation is big.
Solvent Choice for Liquid-Liquid Extraction
I'd mentioned previously that I'd tried extracting with different solvents to compare partition coefficients, but didn't trust my result. I've now repeated that experiment with the P. aquatica extract and I got basically the same result, so I'll report that now.
To each of four microcentrifuge tubes, I added 1 mL of the extract and 200 uL concentrated ammonia. I then added 500 uL of ethyl acetate, dichloromethane, or limonene. I did the limonene twice at two temperatures, 0 C and 50 C, the others once at room temperature. For the limonene samples, the aqueous extract and solvent were both pre-heated or pre-chilled, and I worked quickly whenever they weren't on their respective water baths.
I shook each tube, then left it for about ten minutes to separate. Only the cold limonene formed a clean interface; the others were strong emulsions. I centrifuged to separate, forming three layers, the two desired ones plus a thin waxy one, denser than water but lighter than DCM. This complicated my efforts to syringe off the solvent, but I did the best I could.
I added each solvent to 50 mL 0.25% citric acid in water. The ethyl acetate dissolved, which seemed fine. The DCM partially dissolved and the rest sank to the bottom, which also seemed fine. The limonene formed a cloudy emulsion, which did not seem fine (since I don't want to inject anything into my HPLC that won't dissolve in my eluent, and limonene doesn't much). I centrifuged and the extracts went clear again, and I carefully syringed off some water, avoiding the thin slick of limonene on the top. I injected these under the same chromatographic conditions as before.
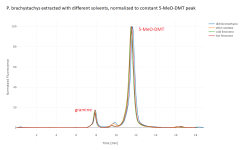
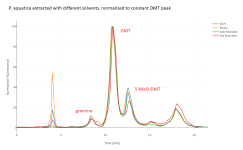
I've superimposed the four chromatograms for the four solvent options, normalized to the same 5-MeO-DMT peak height (for both species) and normalized to the same DMT peak height (for P. aquatica only, since that peak is tiny in the P. brachystachys). We see that for a given yield of 5-MeO-DMT, all the solvents pull almost exactly the same amount of gramine. For a given yield of DMT, the limonene pulls maybe 30% less gramine than the DCM or EtOAc. Temperature makes some difference, but not huge. P. aquatica has many unidentified peaks, and the limonene makes a much bigger difference on some of those, especially when cold. These chromatograms are fluorescence only; there are additional unidentified peaks in the absorption.
I don't think the gramine is particularly harmful under my conditions (yours may differ), but I don't think the limonene helps remove it much.
@endlessness previously
reported excellent rejection of gramine in an extraction using limonene followed by salting as the acetate. I think that must be correct (since there's no obvious experimental error that could result in that chromatogram, and I assume the peaks are correctly identified since it's GC-MS), but that the mechanism is something other than simple insolubility in limonene.
Alkaloid Content When Grown Indoors vs. Outdoors
I've also sampled the P. brachystachys that I germinated, grew, and sampled indoors under lights, then moved outdoors. Indoors, the plants consistently yielded about 950 ug 5-MeO-DMT per g fresh weight. Three samples outdoors yielded 447 ug/g, 428 ug/g, and 406 ug/g. I retook samples of plants from that same batch that I'd left indoors, yielding 562 ug/g, 908 ug/g, 850 ug/g, 1005 ug/g, 1221 ug/g, and 1150 ug/g. I'm not sure what happened with that first low sample indoors. That's the reason why I took so many additional samples, but those were all consistent.
So it seems like growing outdoors will probably yield about half the alkaloids of indoors for me. This may be caused by differences in:
- daily light integral; I've seen claims that alkaloids are higher under shade cloth
- dryback or nutrition; I irrigated daily outdoors with a hydroponic-type fertilizer solution, but I did get more dryback there than indoors; but drought stress is reported to increase alkaloid content, not decrease
- photoperiod; though I'm at 16 hours on my lights and 13 hours sunrise to sunset now, not so different
- temperature; the average outdoors is not that different from indoors now, but there's much more day-night swing.
My indoor space is currently overfull (mostly with cactuses and Sceletium), but I've cleared some room and will grow a square foot or so under lights. I've sown the rest outdoors in partial shade and will compare, and with luck those will yield enough to extract at larger scale.

we'll be posting the TLC results on that thread from now on. See you guys there.