psilocybin
Rising Star
Any questions or problems you may run into during an extraction can be asked to get other members input in this thread.

meino said:Thank You very much,
I'll try not to poison myself.
You forgot to mention the quantity for hot naphtha for recrystallization (I used 100g of root bark).
Wouldn't a syringe be even better than a pipette?
And I also do not use water for recrystallization, correct?
meino said:thanks
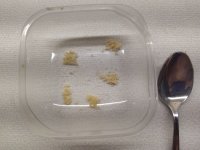
So I got 0,14g crystals now, pretty bad, eh?
I divided them into five pieces do this looks like a correct serving size?
")

dombu said:Using stainless steel tray for evaping instead of glass? is it ok or can it mess things up? I am asking this because I am getting little to no yield from my extractions.
Also about freeze precipitating in a 500ml beaker instead of a pyrex dish, will it affect the crystallization speed, yield?
dombu said:Voidmatrix said:What kind of water are you using in your extraction?
One love
Well its labelled distilled water, guess i have to get some branded distilled water then.
Bruce B. said:I am curious about yield ratios. If say you had 350 g of useful stuff, what is the yield of stuff you will end up with. I am new to this and haven't yet experienced what I know I need desperately. Any help is appreciated , I have Lye and Naphtha. Thank you.
if it's bark particles, a coffee filter should catch them.Jugginlow said:Hello, I am currently preforming my first extraction following Q21Q21’s vinegar/lime a/b tek : 2 (the fluffy white fun fest).
Some background on the extraction, I am using a small amount of MHRB (30g) with calcium hydroxide and naphtha.
I am currently in the middle of my first naphtha pull. My question is what is the stuff rising into the solvent? Is this a sign of emulsion? What is the second liquid layer under the solvent (it is a deep reddish brown color)? Is there any point in collecting that liquid? I’ve been giving it a hot/warm water bath for about an hour now and was going to continue for another hour before attempting to remove the naphtha into my freeze precipitation container (is this the right thing to do?) Any information given will be extremely helpful as I am just a humble novice. I’ve been browsing the forum for the past few days and I believe this is genuine magic glad to join the community! (Is there any way I can upload pictures I’ve taken to help you help me?)
Jugginlow said:Thank you both for the extremely timely reply
Question about the evaporation process : what would be the best method? Is a heat lamp a good choice? How long should I let it evaporate for? And how much will it reduce the solvent by? Also could I preform the evaporation in the same container that I will freeze precipitate from? (Approx 30ml of solvent was used)